01 November 2020: Review Articles
Long-Term Respiratory and Neurological Sequelae of COVID-19
Fuzhou Wang1ABCDEF, Richard M. Kream2ABCDEF, George B. Stefano23ABCDEF*DOI: 10.12659/MSM.928996
Med Sci Monit 2020; 26:e928996






Abstract
ABSTRACT: Since the initial reports of coronavirus disease 2019 (COVID-19) in China in late 2019, infections from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) have spread rapidly, resulting in a global pandemic that has caused millions of deaths. Initially, the large number of infected people required the direction of global healthcare resources to provide supportive care for the acutely ill population in an attempt to reduce mortality. While clinical trials for safe and effective antiviral agents are ongoing, and vaccine development programs are being accelerated, long-term sequelae of SARS-CoV-2 infection have become increasingly recognized and concerning. Although the upper and lower respiratory tracts are the main sites of entry of SARS-CoV-2 into the body, resulting in COVID-19 pneumonia as the most common presentation, acute lung damage may be followed by pulmonary fibrosis and chronic impairment of lung function, with impaired quality of life. Also, increasing reports have shown that SARS-CoV-2 infection involves the central nervous system (CNS) and the peripheral nervous system (PNS) and directly or indirectly damages neurons, leading to long-term neurological sequelae. This review aims to provide an update on the mechanisms involved in the development of the long-term sequelae of SARS-CoV-2 infection in the 3 main areas of lung injury, neuronal injury, and neurodegenerative diseases, including Alzheimer disease, Parkinson disease, and multiple sclerosis, and highlights the need for patient monitoring following the acute stage of infection with SARS-CoV-2 to provide a rationale for the prevention, diagnosis, and management of these potential long-term sequelae.
Keywords: COVID-19, Nervous System, Respiratory System, SARS Virus, severe acute respiratory syndrome, COVID-19, Disease Progression, Lung Injury, Neurodegenerative Diseases, Pandemics, Pulmonary Fibrosis, Quality of Life, SARS-CoV-2, Time Factors
Background
Since the initial reports of coronavirus disease 2019 (COVID-19) in China in late 2019, infections from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) have spread rapidly, causing a global pandemic that has resulted in millions of deaths [1]. While clinical trials for safe and effective antiviral agents are ongoing, and while vaccine development programs are being accelerated, long-term sequelae of SARS-CoV-2 infection have become increasingly recognized and are of increasing concern [2].
The transmission and spread of SARS-CoV-2 did not follow the pattern of other respiratory viruses. Transmission of SARS-CoV-2 increased through the winter of 2019 and 2020, the hot summer of 2020, and into the autumn of 2020. The high rates of airborne and contact spread and persistence of the virus on surfaces explain the rapid spread and the difficulty in controlling infection and the development of COVID-19 [3]. According to the currently available evidence, SARS-CoV-2 can affect every organ in the body, leading to acute organ damage and long-term sequelae, with the latter effects only recently becoming realized [4]. In addition to the high mortality rates from COVID-19 in the elderly and vulnerable, the long-term sequelae from this disease, and the possibility of re-infection due to lack of post-infection immunity, have become major concerns [4–6]. Currently, there are no specific antiviral agents for the treatment of SARS-CoV-2 infection, which mainly relies on supportive care, but there are accelerated programs to develop vaccines that are based on the unique features of the virus, such as the spike protein domains [7].
The most common sites of infection of SARS-CoV-2 are the upper and lower respiratory tracts when the virus is inhaled, and the severity of lung damage is closely related to the severity of infection [8–10]. ‘Post-COVID-19,’ or ‘chronic COVID,’ and the gradual loss of lung function due to pulmonary interstitial fibrosis can have profound effects on daily quality of life for people initially believed to have recovered from COVID-19 [8–10]. In addition, the central nervous system (CNS) and the peripheral nervous system (PNS) are both acutely damaged by SARS-CoV-2 and also show long-term damage [8]. The neuronal damage caused by COVID-19 may be the driving force behind chronic degenerative diseases of the nervous system [8]. Regardless of its direct or indirect effects, damage to the CNS following COVID-19 may be permanent.
This review aims to provide an update on the mechanisms involved in the development of the long-term sequelae of SARS-CoV-2 infection in the 3 main areas of lung injury, neuronal injury, and neurodegenerative diseases, including Alzheimer disease (AD), Parkinson disease (PD), and multiple sclerosis (MS), and highlights the need for patient monitoring following the acute stage of infection with SARS-CoV-2 to provide a rationale for the prevention, diagnosis, and management of these potential long-term sequelae.
Lung Injury Due to SARS-CoV-2
The respiratory system is the front-line of the SARS-CoV-2 infection [9,10]. The virus can injure the lungs in 3 ways: acute respiratory distress syndrome (ARDS) with diffuse alveolar damage (DAD), diffuse thrombotic alveolar microvascular occlusion, and inflammatory mediator-associated airway inflammation [9,10]. The results of these combined actions include impaired alveolar oxygenation, hypoxemia, and acidosis. In the absence of effective treatment, the consequences of this poor oxygenation are either death of the patient from respiratory failure, or the sequelae of permanent lung injury if the patient recovers [9–11].
Lung Alveolar Injury and Diffuse Alveolar Damage (DAD) in Acute Respiratory Distress Syndrome (ARDS)
SARS-CoV-2 directly attacks type 2 pneumocytes by binding to the angiotensin-converting enzyme 2 (ACE2) receptor on the cell surface and destroys them [4]. The degree of cell damage may depend not only on the effects of viral replication, but also on the release of pro-inflammatory cytokines, resulting in impaired function of type 2 pneumocytes [4,10]. These 2 effects result in impaired cell function, followed by cell death (necrosis) or apoptosis, exudates, desquamation of pneumocytes, and formation of hyaline membranes, which are characteristic of diffuse alveolar damage (DAD) [10]. Interstitial edema and inflammatory infiltrates of mononuclear and multinucleated syncytial cells also contribute to alveolar dysfunction [10–12]. As a result, the typical pathophysiological feature of COVID-19 pneumonia/ARDS is that alveolar gas exchange and oxygenation are severely impaired [13].
Thrombosis of the Alveolar Microcirculation Due to Coagulopathy
Effective alveolar gas exchange by diffusion depends on the integrity and normal function of the alveolar epithelium and a normal microcirculation with patent alveolar capillaries [13,14]. SARS-CoV-2 damages the 2 major functional components of alveolar gas exchange: the integrity of the alveolar epithelium, and the patency and function of the alveolar microcirculation [15,16]. The ACE2 receptor facilitates entry of SARS-CoV-2 into cells and also serves as a ‘bridge’ for the virus to directly target vascular endothelial cells, which results in endothelial cell activation [4]. Combined with the release of pro-inflammatory cytokines and chemokines, activated endothelial cells show upregulation of von Willebrand factor (vWF) and adhesion molecules, including intercellular adhesion molecule (ICAM)-1, P-selectin, and E-selectin [12,16]. These changes are followed by the aggregation of platelets and leukocytes and activation of the complement system [16]. Therefore, neutrophil extracellular traps (NETS) that activate the direct contact pathway for thrombosis are released, accompanied by complement activation that induces the release of tissue factor (TF) [16]. In addition, endothelial cell hypoxia-inducible factors (HIFs) upregulate TF, and platelet aggregates result in thrombus formation [16].
Airway Injury Due to Pro-Inflammatory Cytokines
SARS-CoV-2 not only damages alveolar gas diffusion functions, but also induces airway inflammation to reduce the ventilation function of the airway. The common association of bronchopneumonia with COVID-19 pneumonia provides direct evidence that SARS-CoV-2 affects airway ventilation function [8,9]. Reports of inflammatory infiltration of bronchioles, bronchi, and trachea [14–17], together with the increased inflammation on scintigraphy of the upper respiratory tract in non-smokers with COVID-19, supports the direct damage done by SARS-CoV-2 to the airway [18].
Long-Term Sequelae of COVID-19 Pneumonia
Because COVID-19 and the SARS-CoV-2 pandemic has been ongoing for less than 1 year, it is difficult to identify and investigate the long-term sequelae of SARS-CoV-2 infection, although some are now becoming apparent. However, some results have been reported from follow-up of patients who have recovered from SARS-CoV-1 infection in 2003, which may indicate what to expect in the long term from SARS-CoV-2 infection [17,18]. Because SARS-CoV-2 has increased pathogenicity and invasiveness and has now infected millions of people worldwide, it is important to attempt to make early predictions of the possible long-term sequelae of COVID-19 and to formulate relevant prevention and intervention strategies.
In 2003, more than 8000 cases and 900 deaths resulted from SARS-CoV-1 infection worldwide. In a follow-up study that enrolled 97 SARS-CoV-1 survivors, 1-year follow-up identified abnormalities on chest X-ray in 28% of patients, the severity lung abnormalities on imaging was closely related to the extent of functional lung impairment, and the overall quality of life in SARS-CoV-1 survivors was worse than that of an age-matched comparison group [17]. A study that followed up SARS-CoV-1 survivors for 2 years [18] and another for 15 years [19] showed similar results. Lung ventilation function of all follow-up patients had varying degrees of damage, and the lung diffusion function in more than one-third was significantly impaired [17–19]. The typical pulmonary fibrotic lesions on lung computed tomography (CT) scan included air trapping, ground-glass opacities (GGOs), reticulation, and traction bronchiectasis [20]. Intralobular and interlobular septal thickening were also common among SARS-CoV-1 survivors, suggesting that the lungs underwent self-healing and continuous remodelling after severe damage [21]. The fibrotic changes that occur after severe lung injury are an essential manifestation of the ability of the lungs to repair and remodel. Based on the findings from these previous studies on patients with SARS-CoV-1, approximately one-third of the survivors had significant pulmonary fibrosis [18,19].
According to data from the World Health Organization (WHO), by September 23, 2020, the total number of COVID-19 cases worldwide reached 31 664 104 [22]. After subtracting the number of deaths, a conservative calculation indicates that one-third of the survivors who have been infected with SARS-CoV-2 will develop significant pulmonary fibrosis, and the number who may develop chronic sequelae of pulmonary fibrosis will reach an estimated 10 230 628 [22]. In addition, the COVID-19 pandemic has not ended, and the number of people infected with SARS-CoV-2 increases daily. If this pandemic continues, the number of survivors with chronic lung fibrosis after SARS-CoV-1 infection will increase further.
The evolution of pulmonary fibrosis shows the potential of the lungs to continue to heal after being severely injured [23], from the early stages of pulmonary edema, desquamation of pneumocytes, the formation of hyaline membranes or DAD, inflammation, and organization, to the late stages of lung repair involving fibrosis and interstitial remodelling with impaired gas diffusion [23]. Following SARS-CoV-2 infection, and in a similar way to the effects reported from SARS-CoV-1 infection, the formation of intra-alveolar thrombosis and airway inflammatory viral damage further contribute to the development of pulmonary fibrosis [23–25]. SARS-CoV-2 can induce pulmonary fibrosis by promoting the upregulation of pro-fibrotic signaling molecules, including transforming growth factor-beta (TGF-β) [24,25]. The nucleocapsid protein of SARS-CoV-1 can directly promote the upregulation of TGF-β expression [24–27]. However, SARS-CoV-1 can reduce angiotensin II (Ang II) clearance by reducing ACE2, and Ang II can induce TGF-β expression [25]. The final result of these 2 effects is TGF-β-mediated pulmonary fibrosis. Considering that the similarity of nucleocapsid protein between SARS-CoV-2 and SARS-CoV-1 is as high as 90% [26,27], it is possible to speculate that SARS-CoV-2 will also have a similar molecular mechanism. The proposed long-term effects of SARS-CoV-2 infection in the respiratory tract are shown in Figure 1.
Given the potential risks of SARS-CoV-2-induced pulmonary fibrosis, it is important to prepare and implement early and effective preventive measures for COVID-19 survivors [18]. Approaches include the effective prevention of the spread of SARS-CoV-2, inhibition of virus replication, blocking the inflammatory response, and treatment to prevent pulmonary fibrosis at an early stage. These 4 approaches may prove important strategies to achieve effective prevention of pulmonary fibrosis as a long-term consequence of COVID-19 pneumonia.
Neuronal Injury Due to SARS-CoV-2
With the rapid increase in the numbers of COVID-19 patients and the appearance of different symptoms and signs, reports of neurological damage have gradually attracted attention [28,29]. It was initially thought that SARS-CoV-2 had great difficulty in passing through the dense blood–brain barrier (BBB), but this is not the case. Post-mortem studies on the cerebral pathology of COVID-19 patients and the use of an advanced 3D microfluid model of the human BBB identified 3 important findings [29,30]. First, the SARS-CoV-2 spike (S) protein-binding receptor, ACE2, is widely expressed in brain microvascular endothelial cells [29,30]. Second, the S protein can directly damage the integrity of the BBB to varying degrees [29,30]. Third, the S protein can induce the inflammatory response of microvascular endothelial cells that change the function of the BBB [29,30]. These findings support that SARS-CoV-2 can alter the BBB and enter the brain, and support the appearance of neurological symptoms, the formation of fatal microthrombi, and even the occurrence of encephalitis associated with COVID-19 [29,30]. These neurologic associations of COVID-19 support the clinical reports of early neurological changes, and support the potential basis for the occurrence of long-term neurological sequelae. In addition, to cross the BBB, SARS-CoV-2 may enter the brain by trans-synaptic transfer, optic and olfactory nerve channels, and vascular endothelial cells [29,30].
Indirect Evidence of Damage to the CNS by SARS-CoV-2
The SARS-CoV-1 infection epidemic in 2003 was reported to cause various types of neurological damage, including the axonal-variant Guillain-Barré syndrome, ischemic stroke, and seizures [30–32]. Studies confirmed the direct replication of SARS-CoV-1 in brain tissue, and the pathological anatomy of the brains of patients with SARS-CoV-1 showed that the virus was present in the cytoplasm of cortical and hypothalamic neurons [33]. The pathological changes of brain tissue in patients with encephalitis caused by SARS-CoV-1 included neuronal necrosis, glial cell hyperplasia, and infiltration of monocytes and T cells [34]. SARS-CoV-1 was also shown to directly cause the death of cerebral neurons in ACE2 transgenic mice, but without the pathological changes of encephalitis [35]. These results provide indirect evidence for possible mechanisms of cerebral damage caused by SARS-CoV-2.
Neuro-Invasive Mechanisms of SARS-CoV-2
For both SARS-CoV-1 and SARS-CoV-2, these highly pathogenic coronaviruses enter the brain via the viral S protein, which can bind to the ACE2 receptor [4]. The expression of the ACE2 receptor in the cell determines the cell tropism of the virus [4,35–37]. Increasing numbers of studies have supported this molecular pathogenesis in the brain, as the distribution of ACE2 expression at different sites in animal and human brain tissues is widely expressed in neurons, astrocytes, and oligodendrocytes throughout the brain [36]. These findings support a molecular mechanism by which SARS-CoV-2 enters the brain and then causes neuronal damage [36–38].
Olfactory Nerve Channels and Trans-Synaptic Viral Transfer
The olfactory nerve is now considered to be a potential pathway for entry of the SARS-CoV-2 into the brain [7,36], as sustentacular cells that maintain the integrity of the sense of smell [37] and stem cells in the olfactory epithelium [38] all highly express ACE2 and transmembrane serine protease 2 (TMPRSS2) [37]. The entry proteins ACE2 and TMPRSS2 may have a role in the binding of SARS-CoV-2 to the olfactory bulb and sustentacular cells, followed by travel into the brain [38,39]. Nicotinic neuronal processes also have been implicated [7]. Previous studies have shown that several viruses can enter the brain through olfactory neurons, including SARS-CoV-1 [39], MERS-CoV [40,41], and HCoV-OCR43 [41]. These previous studies also provide indirect evidence that SARS-CoV-2 can enter the brain through the olfactory pathway.
The expression of ACE2 on the neuronal membrane and cytoplasm also provides a mechanism for the trans-synaptic transfer of SARS-CoV-2 [42]. In ACE2 transgenic mice, SARS-CoV-1 was reported to rapidly enter the brain through the olfactory bulb, to transfer rapidly trans-synaptically, and directly destroy cerebral neurons without causing inflammatory infiltration [35]. In addition to SARS-CoV-1, the properties of trans-synaptic transfer have also been confirmed in other viruses, such as HCoV-OC43, hemagglutinating encephalomyelitis virus 67 (HEV67), and avian bronchitis virus [43]. When the SARS-CoV-2 virus binds to the ACE2 receptor, it travels in a retrograde axonal manner to reach the central nervous system [44]. When the virus reaches the synaptic cleft, membrane coating-mediated exocytosis and endocytosis and vesicle transport result in the trans-synaptic transfer of the virus from neuron to neuron and from neuron to satellite cell [44,45]. In addition, the rapid retrograde or anterograde intracellular axonal transport that relies on microtubules provides a structural basis for further virus transfer [43,46].
Vascular Endothelial Cells of the Blood–Brain Barrier (BBB) and Immune Cells
The BBB is considered to be another pathway by which SARS-CoV-2 enters the brain [47,48]. There are 2 ways for the virus to cross the BBB: the vascular endothelial cell pathway and the immune cell pathway [29]. Vascular endothelial cells regulate the permeability of BBB through tight junctions [29]. The extensive expression of ACE2 in vascular endothelial cells throughout the body provides the molecular mechanism by which SARS-CoV-2 penetrates the BBB and invades the brain [29]. Electron microscopy images have shown that SARS-CoV-2 binds to the ACE2 receptor and enters vascular endothelial cells by endocytosis and exocytosis, thereby achieving cell-cell transfer of the virus [47]. However, the virus does not replicate during the process of trans-endothelial cell transfer, but viral replication is delayed until it reaches its target cells, such as neurons, glia, and vessels, and binds to ACE2 before starting to replicate [48].
In addition to targeting the vascular endothelium, immune cells may function as another pathway for SARS-CoV-2 to cross the BBB [49]. This approach is termed a ‘Trojan horse’ mechanism because it requires at least 2 conditions: immune cells that express ACE2, and conditions in which SARS-CoV-2 does not replicate [49]. Immune cells, including lymphocytes, granulocytes, and monocytes, all highly express ACE2 [50–53]. Recently, Abassi et al. [54] showed that SARS-CoV-2 can enter the cytoplasm of macrophages by binding to ACE2 on the surface. The virus-containing macrophages were functioning using a ‘Trojan horse’ mechanism to migrate to other locations like the CNS for replication [54]. The findings from this recent study suggest that the infected immune cells are another pathway that carries SARS-CoV-2 across the BBB and into the brain [54]. Although these findings require support from further studies, there is the possibility that SARS-CoV-2 can use cells of the immune system to spread throughout the body and to cross the BBB, a process much like the human immunodeficiency virus.
Underlying Molecular Mechanisms of SARS-CoV-2-Related Neuronal Injury
No matter what method of invasion SARS-CoV-2 uses, when it finally reaches its destination, it rapidly replicates and then uses its unique mechanisms to cause cell death or functional impairment [50]. Rapid viral replication, direct cell damage, and activation of the immune system and inflammatory mediators, including cytokines, are the likely causes of the acute symptoms of COVID-19 and may explain the long-term sequelae of SARS-CoV-2 infection.
Pro-Inflammatory Cytokines and the ‘Cytokine Storm’
The systemic increase in inflammatory mediators, now termed the ‘cytokine storm,’ may explain the multi-organ damage found in some patients with COVID-19 and may also explain the effects of SARS-CoV-2 on the CNS [55]. The release of a large number of pro-inflammatory cytokines increases vascular permeability, abnormal blood coagulation, and multiple-organ failure [55]. These cytokines may also have a role in increasing microvascular permeability in the CNS, facilitating the entry of SARS-CoV-2 through the BBB and into the brain [28,29]. The ‘cytokine storm’ may also promote the formation of microthrombi by activating the coagulation system [55,56]. Brain-imaging findings in patients COVID-19 with neurological involvement have shown neuroradiological patterns in the medial temporal lobe, multifocal lesions in the cerebral white matter, and microhemorrhages [57]. Current brain-imaging methods include use of fluid-attenuated inversion recovery (FLAIR) cerebral magnetic resonance imaging (MRI) [57].
Mitochondrial Pathways
Mitochondria are key cell organelles that maintain normal cell function and cell dynamics and maintain cellular homeostasis. Functional impairment of cell mitochondria results in cell apoptosis, necrosis, or dysfunction. SARS-CoV-2 infection results in organ damage at the cellular level in several ways. The SARS-CoV-2 RNA genome and all sub-genomic RNAs integrate into the host mitochondrial matrix, resulting in a viral-mitochondrial interaction that leads to virus replication and increased vital load, and SARS-CoV-2 RNA transcripts in cell mitochondria ‘hijack’ mitochondrial function to suppress the immune response and promote virus replication [58–60]. Eventually, the infected cells, including neurons, may undergo necrosis, apoptosis, or dysfunction due to oxidative stress and calcium ion influx, with impaired mitochondrial function [61,62]. Although necrosis and apoptosis of infected host cells may seem to reduce the survival of the virus, these effects result in damage to the CNS. If the host survives, the virus will be shed and be transmitted to another host, and chronic neurological damage is more beneficial to the virus than is death of the host [63–65]. This explanation of the long-term pathogenesis of cerebral SARS-CoV-2 infection may also explain the long-term neurological sequelae.
Autophagy
Given the causal interaction between autophagy and apoptosis, the possible role of SARS-CoV-2 between them seems more complicated. To achieve maximal replication and spread, viruses have formed a very special survival law. The virus maintains the integrity of the host cells infected by the virus in the incubation period by unique means, further prevents the initiation of the autophagy and apoptosis program of the infected host cells, and finally provides a guarantee for the release of a greater number of progeny virions [65,66]. Paradoxically, the virus does not actively induce autophagy and apoptosis with the purpose of using the remnants of cell destruction as vehicles for further propagation or as a strategy to avoid immune attacks [65]. Although the relationship between SARS-CoV-2 and autophagy currently remains unclear, possible mutually beneficial interactions cannot be completely ruled out [65].
Autophagy and apoptosis occur in the fierce battle between the virus and the host. Infected host cells gather a large number of autophagosomes to activate autophagy-linked apoptosis, aiming to cut off the loop of virus replication [63,66]. Therefore, it is speculated that the virus does its best to delay the formation and aggregation of autophagosomes in the infected host cells at the beginning of infection and to use the time available to replicate. However, as the infection continues and the number of autophagosomes increases, the virus turns to promoting the accelerated formation and aggregation of autophagosomes to activate the apoptotic program, thereby facilitating better apoptotic use of cell remnants as carriers to accelerate viral spread. Further studies on the role of apoptosis and autophagy in the pathogenesis may have implications for the development of treatments for acute and chronic neurological sequelae [67,68]. The proposed long-term neurological effects of SARS-CoV-2 infection are shown in Figure 2.
Neurodegenerative Diseases and SARS-CoV-2
Neurodegenerative disease is an umbrella concept including a range of conditions that primarily affect the neurons in the human brain and is one of the key factors leading to the decline in quality of life. Whether SARS-CoV-2 causes neurodegenerative diseases or accelerates their premature occurrence is still unclear, and it is also difficult to draw conclusions within just a few months. However, the high expression of the ACE2 receptor in a wide range of sites in the brain not only provides an initial target for SARS-CoV-2 to cause acute brain damage, it may also be the basis for later neurodegenerative changes [68]. This possibility is supported by the findings from recent studies that showed the presence of functional inhibition of viral and nicotinic acetylcholine receptor complexes in the pathogenesis of SARS-CoV-2 infection [7].
Alzheimer Disease (AD)
In addition to age, there are many other important risk factors for Alzheimer disease (AD) [69]. Recently published studies have focussed on the potential causal relationship between viral infections and AD [70]. Given the identification of damage to the CNS by SARS-CoV-2, there is concern regarding its long-term effects on cognitive function [71,72]. It is possible that further long-term studies will be required to identify the relationships among SARS-CoV-2 infection, AD, and other neurodegenerative sequelae. Neuroinflammatory responses, synaptic pruning, and neuronal loss are the structural basis of AD [73], and SARS-CoV-2 infection most likely accelerates these processes. The excitotoxic reaction caused by the imbalance between glutamatergic and GABAergic responses is a potential mechanism that promotes neuronal loss and further cerebral tissue damage [74]. The simultaneous expression of ACE2 in glutamatergic and GABAergic neurons indicates that SARS-CoV-2 infection can affect the balance of both signaling pathways in the CNS [36, 68]. Furthermore, the trans-synaptic transfer and the axonal retrograde or anterograde movement of SARS-CoV-2 make it possible for the virus to slowly and diffusely infiltrate the entire brain, and it also promotes the chronicity and the neurodegenerative changes months and years after the acute infection [42,44,45].
Parkinson Disease (PD)
Compared with AD, the potential CNS damage that is localized to the substantia nigra striatum that can result in Parkinson disease (PD) seems to be more limited [75]. However, several recent studies have shown that patients with PD not only show motor dysfunction, their cognitive and memory functions are also severely impaired [76–78]. Also, the pathogenesis of PD is associated with neuroinflammation, synaptic pruning, and neuron loss [75], sharing commonalities with AD [79,80]. However, in PD, different CNS sites are damaged, with different types of neurons being more severely affected [81]. Currently, although there is no direct evidence that SARS-CoV-2 causes or accelerates PD [82], it should be noted that the wide expression of ACE2 at different areas in the CNS provides a molecular basis for SARS-CoV-2 to mediate or accelerate the occurrence of PD.
There is currently limited evidence from clinical studies to support an association between the onset of PD as a late complication of SARS-CoV-2 infection. Hyposmia and anosmia are also precursor clinical symptoms of PD [83], and are early symptoms in COVID-19 patients, occurring without nasal congestion and rhinorrhea [84–87]. A recently published case report described a patient with COVID-19 who had symptoms of encephalopathy with imaging changes of rare bilateral basal ganglia hemorrhagic injury [87]. Therefore, for AD and other neurodegenerative conditions, more long-term studies are required to identify the relationship between SARS-CoV-2 infection and PD.
Multiple Sclerosis (MS)
Multiple sclerosis (MS) is associated with focal gray and white matter demyelination and diffuse neurodegeneration of the brain caused by inflammation [88]. Current knowledge of the neurological changes caused by SARS-CoV-2 shows some similarities with those found in MS. First, the pro-inflammatory ‘cytokine storm’ caused by SARS-CoV-2 infection is the initiating factor of CNS neuroinflammatory damage [89]. Second, SARS-CoV-2 can cause demyelination in the brain and spinal cord [90]. A recently published case report showed that SARS-CoV-2 infection was associated with signs and symptoms similar to those of MS [91]. Previous studies have shown an association between coronavirus infection and the onset of MS [92]. If an association between SARS-CoV-2 infection and demyelinating neurological disease does exist, this will result in a therapeutic dilemma, as immunotherapy is used to treat these diseases, including MS [93]. More long-term studies will be required to identify the relationship between SARS-CoV-2 infection and MS.
Conclusions
As of October 2020, COVID-19, due to infection with SARS-CoV-2, has become a global pandemic less than 1 year following the identification of the first cases in Wuhan, China. This review has provided an update on the current status of the mechanisms involved in the long-term sequelae of SARS-CoV-2 infection in the 3 main areas of lung injury, neuronal injury, and neurodegenerative disease. Clinical studies, laboratory studies, clinical trials of potential therapeutic agents, and vaccine development programs continue to accelerate. The SARS-CoV-2 virus has many unique properties that increase its transmission and pathogenic effects. At such an early stage of the pandemic, the potential long-term sequelae of COVID-19 are just beginning to be realized. This review has highlighted the need for more long-term clinical follow-up data on patients who have had COVID-19, and for attention to the management of long-term sequelae, which will emerge in patient care settings. Finally, the economic impact of this disorder, together with patient care, must be worked out in advance.
Figures
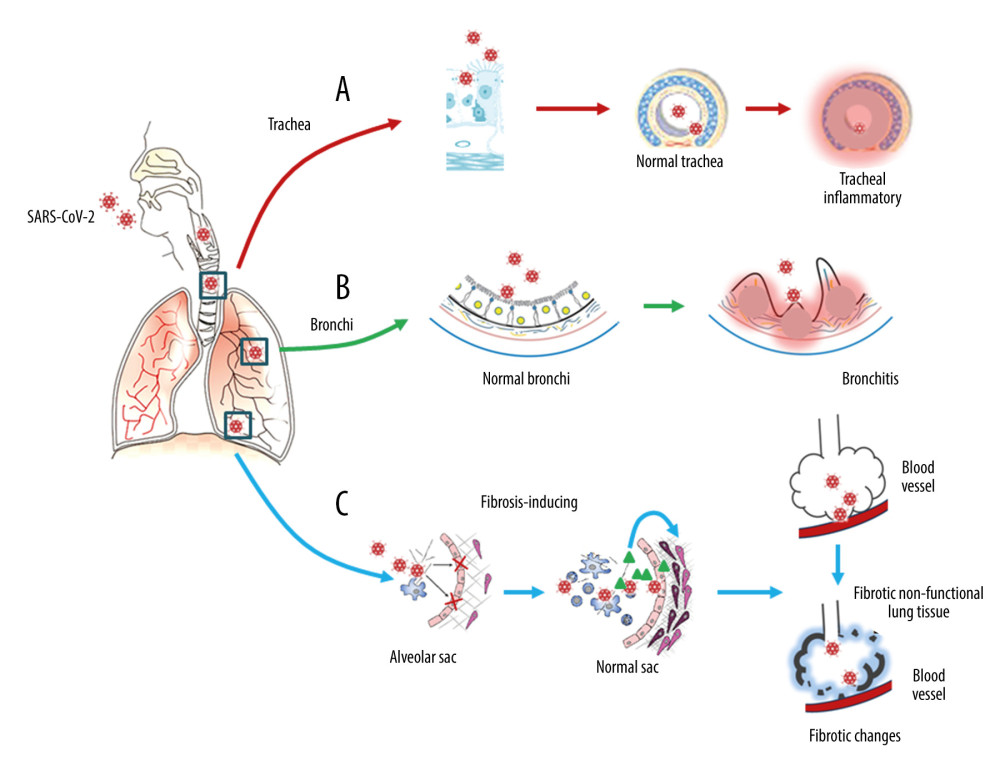 Figure 1. Respiratory injury associated with SARS-CoV-2 infection. SARS-CoV-2 induces inflammatory changes in all 3 main locations of the respiratory system from the trachea (A), to the bronchi (B), and the alveolar sacs (C). The respiratory system will experience acute inflammation following the pro-inflammatory ‘cytokine storm,’ followed by long-term fibrotic changes. Pulmonary fibrosis is associated with the upregulation of transforming growth factor-beta (TGF-β) and chronic inflammation.
Figure 1. Respiratory injury associated with SARS-CoV-2 infection. SARS-CoV-2 induces inflammatory changes in all 3 main locations of the respiratory system from the trachea (A), to the bronchi (B), and the alveolar sacs (C). The respiratory system will experience acute inflammation following the pro-inflammatory ‘cytokine storm,’ followed by long-term fibrotic changes. Pulmonary fibrosis is associated with the upregulation of transforming growth factor-beta (TGF-β) and chronic inflammation. 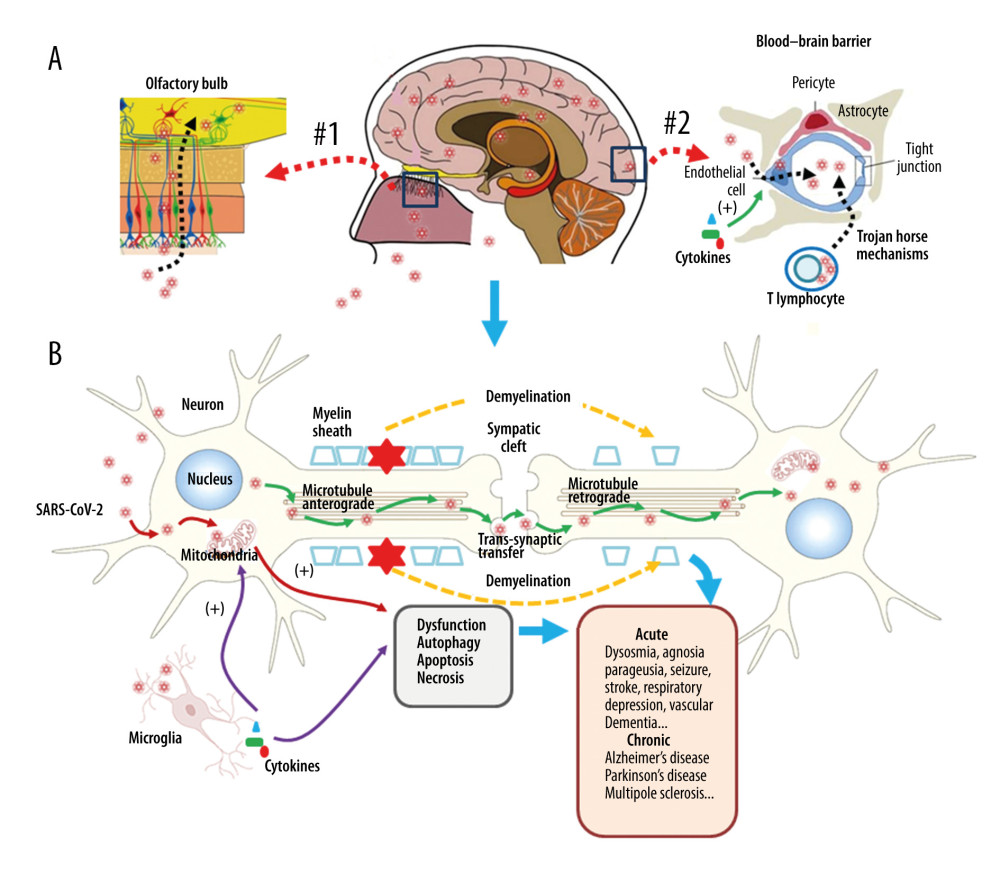 Figure 2. Neuronal injury associated with SARS-CoV-2 infection. SARS-CoV-2 enters into the central nervous system (CNS) through 2 major routes: the olfactory pathway (#1 in A), and the blood–brain barrier (BBB) pathway (#2 in A). The virus can migrate into the CNS directly by endocytosis with the assistance of inflammatory cytokine-induced increased vascular permeability and indirect transfer via a ‘Trojan horse’ mechanism. (B) After binding to its membrane receptor, ACE2, SARS-CoV-2 will be engulfed into neuronal cytosol and move to connect with cytosol-located angiotensin-converting enzyme 2 (ACE2). The viral RNAs enter the mitochondria or form into autophagolysosomes to initiate autophagy and/or apoptosis. Microglia and immune cells produce pro-inflammatory cytokines, which result in further abnormalities in mitochondrial function. When the virus enters the neurons, it combines with the axonal microtubules with anterograde and retrograde spread to the synapse and enters the next level of neurons by trans-synaptic transfer and endocytosis. Both the virus and the ‘cytokine storm’ can destroy the myelin sheath of neurons, resulting in acute and chronic neuropathology.
Figure 2. Neuronal injury associated with SARS-CoV-2 infection. SARS-CoV-2 enters into the central nervous system (CNS) through 2 major routes: the olfactory pathway (#1 in A), and the blood–brain barrier (BBB) pathway (#2 in A). The virus can migrate into the CNS directly by endocytosis with the assistance of inflammatory cytokine-induced increased vascular permeability and indirect transfer via a ‘Trojan horse’ mechanism. (B) After binding to its membrane receptor, ACE2, SARS-CoV-2 will be engulfed into neuronal cytosol and move to connect with cytosol-located angiotensin-converting enzyme 2 (ACE2). The viral RNAs enter the mitochondria or form into autophagolysosomes to initiate autophagy and/or apoptosis. Microglia and immune cells produce pro-inflammatory cytokines, which result in further abnormalities in mitochondrial function. When the virus enters the neurons, it combines with the axonal microtubules with anterograde and retrograde spread to the synapse and enters the next level of neurons by trans-synaptic transfer and endocytosis. Both the virus and the ‘cytokine storm’ can destroy the myelin sheath of neurons, resulting in acute and chronic neuropathology. References
1. Kannan S, Shaik Syed Ali P, Sheeza A, Hemalatha K, COVID-19 (Novel Coronavirus 2019) – recent trends: Eur Rev Med Pharmacol Sci, 2020; 24(4); 2006-11
2. Wang F, Kream RM, Stefano GB, An evidence-based perspective on mRNA-SARS-CoV-2 vaccine development: Med Sci Monitm, 2020; 26; e924700
3. Oldfield E, Malwal SR, COVID-19 and other pandemics: How might they be prevented?: ACS Infect Dis, 2020; 6(7); 1563-66
4. Bourgonje AR, Abdulle AE, Timens W, Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19): J Pathol, 2020; 251(3); 228-48
5. Helms J, Tacquard C, Severac FCRICS TRIGGERSEP Group (Clinical Research in Intensive Care and Sepsis Trial Group for Global Evaluation and Research in Sepsis), High risk of thrombosis in patients with severe SARS-CoV-2 infection: A multicenter prospective cohort study: Intensive Care Med, 2020; 46(6); 1089-98
6. George PM, Wells AU, Jenkins RG, Pulmonary fibrosis and COVID-19: The potential role for antifibrotic therapy: Lancet Respir Med, 2020; 8(8); 807-15
7. Stefano ML, Kream RM, Stefano GB, A novel vaccine employing non-replicating rabies virus expressing chimeric SARS-CoV-2 spike protein domains: functional inhibition of viral/nicotinic acetylcholine receptor complexes: Med Sci Monit, 2020; 26; e926016
8. Singal CMS, Jaiswal P, Seth P, SARS-CoV-2, more than a respiratory virus: Its potential role in neuropathogenesis: ACS Chem Neurosci, 2020; 11(13); 1887-99
9. Calabrese F, Pezzuto F, Fortarezza F, Pulmonary pathology and COVID-19: Lessons from autopsy. The experience of European Pulmonary Pathologists: Virchows Arch, 2020; 477(3); 359-72
10. Tian S, Xiong Y, Liu H, Pathological study of the 2019 novel coronavirus disease (COVID-19) through post-mortem core biopsies: Mod Pathol, 2020; 33; 1007-14
11. Xu Z, Shi L, Wang Y, Pathological findings of COVID-19 associated with acute respiratory distress syndrome: Lancet Respir Med, 2020; 8; 420-22
12. Stefano GB, Esch T, Kream RM, Potential immunoregulatory and antiviral/SARS-CoV-2 activities of nitric oxide: Med Sci Monit, 2020; 26; e925679
13. Shao C, Liu H, Meng L, Evolution of severe acute respiratory syndrome coronavirus 2 RNA test results in a patient with fatal coronavirus disease 2019: A case report: Hum Pathol, 2020; 101; 82-88
14. Hsia CC, Hyde DM, Weibel ER, Lung structure and the intrinsic challenges of gas exchange: Compr Physiol, 2016; 6(2); 827-95
15. Miesbach W, Makris M, COVID-19: Coagulopathy, risk of thrombosis, and the rationale for anticoagulation: Clin Appl Thromb Hemost, 2020; 26 1076029620938149
16. McFadyen JD, Stevens H, Peter K, The Emerging threat of (Micro)thrombosis in COVID-19 and its therapeutic implications: Circ Res, 2020; 127(4); 571-87
17. Hui DS, Wong KT, Ko FW, The 1-year impact of severe acute respiratory syndrome on pulmonary function, exercise capacity, and quality of life in a cohort of survivors: Chest, 2005; 128; 2247-61
18. Ngai JC, Ko FW, Ng SS, The long-term impact of severe acute respiratory syndrome on pulmonary function, exercise capacity and health status: Respirology, 2010; 15; 543-50
19. Zhang P, Li J, Liu H, Long-term bone and lung consequences associated with hospital-acquired severe acute respiratory syndrome: A 15-year follow-up from a prospective cohort study: Bone Res, 2020; 8; 8
20. Chang YC, Yu CJ, Chang SC, Pulmonary sequelae in convalescent patients after severe acute respiratory syndrome: Evaluation with thin-section CT: Radiology, 2005; 236; 1067-75
21. Wu X, Dong D, Ma D, Thin-section computed tomography manifestations during convalescence and long-term follow-up of patients with severe acute respiratory syndrome (SARS): Med Sci Monit, 2016; 22; 2793-99
22. World Health Organization: WHO Coronavirus Disease (COVID-19) Dashboard https://covid19.who.int
23. Cheung OY, Chan JWM, Ng CK, The spectrum of pathological changes in severe acute respiratory syndrome (SARS): Histopathology, 2004; 45; 119-24
24. Zhao X, Nicholls JM, Chen Y-G, Severe acute respiratory syndrome-associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor-beta signaling: J Biol Chem, 2008; 283; 3272-3280
25. Zuo W, Zhao X, Chen YG, SARS coronavirus and lung fibrosis: Molecular Biology of the SARS-Coronavirus, 2010; 247-58, Berlin and Heidelberg, Springer
26. Malik YA, Properties of Coronavirus and SARS-CoV-2: Malays J Pathol, 2020; 42(1); 3-11
27. Chang CK, Hou MH, Chang CF, The SARS coronavirus nucleocapsid protein-forms and functions: Antiviral Res, 2014; 103; 39-50
28. Chiappelli F, Towards neuro-CoViD-19: Bioinformation, 2020; 16(4); 288-92
29. Pezzini A, Padovani A, Lifting the mask on neurological manifestations of COVID-19: Nat Rev Neurol, 2020; 24; 1-9
30. Tsai LK, Hsieh ST, Chang YC, Neurological manifestations in severe acute respiratory syndrome: Acta Neurol Taiwan, 2005; 14(3); 113-19
31. Tsai LK, Hsieh ST, Chao CC, Neuromuscular disorders in severe acute respiratory syndrome: Arch Neurol, 2004; 61(11); 1669-73
32. Lau KK, Yu WC, Chu CM, Possible central nervous system infection by SARS coronavirus: Emerg Infect Dis, 2004; 10(2); 342-44
33. Ding Y, Wang H, Shen H, The clinical pathology of severe acute respiratory syndrome (SARS): A report from China: J Pathol, 2003; 200(3); 282-89
34. Xu J, Zhong S, Liu J, Detection of severe acute respiratory syndrome coronavirus in the brain: Potential role of the chemokine Mig in pathogenesis: Clin Infect Dis, 2005; 41(8); 1089-96
35. Netland J, Meyerholz DK, Moore S, Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2: J Virol, 2008; 82(15); 7264-75
36. Barrantes FJ, Central nervous system targets and routes for SARS-CoV-2: Current views and new hypotheses: ACS Chem Neurosci, 2020; 11(18); 2793-803
37. Bilinska K, Jakubowska P, Von Bartheld CS, Butowt R, Expression of the SARS-CoV-2 entry proteins, ACE2 and TMPRSS2, in cells of the olfactory epithelium: Identification of cell types and trends with age: ACS Chem Neurosci, 2020; 11(11); 1555-62
38. Brann DH, Tsukahara T, Weinreb C, Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia: Sci Adv, 2020; 6(31); eabc5801
39. McCray PB, Pewe L, Wohlford-Lenane C, Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus: J Virol, 2007; 81(2); 813-21
40. Jacomy H, Talbot PJ, Vacuolating encephalitis in mice infected by human coronavirus OC43: Virology, 2003; 315(1); 20-33
41. Li K, Wohlford-Lenane C, Perlman S, Middle East respiratory syndrome coronavirus causes multiple organ damage and lethal disease in mice transgenic for human dipeptidyl peptidase 4: J Infect Dis, 2016; 213(5); 712-22
42. Xiao L, Haack KK, Zucker IH, Angiotensin II regulates ACE and ACE2 in neurons through p38 mitogen-activated protein kinase and extracellular signal-regulated kinase 1/2 signaling: Am J Physiol Cell Physiol, 2013; 304(11); C1073-79
43. Dubé M, Le Coupanec A, Wong AHM, Axonal transport enables neuron-to-neuron propagation of human Coronavirus OC43: J Virol, 2018; 92(17); e00404-18
44. Li YC, Bai WZ, Hashikawa T, The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients: J Med Virol; 92(6); 552-55 202
45. Li YC, Bai WZ, Hirano N, Coronavirus infection of rat dorsal root ganglia: ultrastructural characterization of viral replication, transfer, and the early response of satellite cells: Virus Res, 2012; 163(2); 628-35
46. Berth SH, Leopold PL, Morfini GN, Virus-induced neuronal dysfunction and degeneration: Front Biosci (Landmark Ed), 2009; 14; 5239-59
47. Paniz-Mondolfi A, Bryce C, Grimes Z, Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): J Med Virol, 2020; 92(7); 699-702
48. Baig AM, Khaleeq A, Ali U, Syeda H, Evidence of the COVID-19 virus targeting the CNS: Tissue distribution, host-virus interaction, and proposed neurotropic mechanisms: ACS Chem Neurosci, 2020; 11(7); 995-98
49. Desforges M, Le Coupanec A, Brison E, Neuroinvasive and neurotropic human respiratory coronaviruses: potential neurovirulent agents in humans: Adv Exp Med Biol, 2014; 807; 75-96
50. Gu J, Gong E, Zhang B, Multiple organ infection and the pathogenesis of SARS: J Exp Med, 2005; 202(3); 415-24
51. Nicholls JM, Butany J, Poon LL, Time course and cellular localization of SARS-CoV nucleoprotein and RNA in lungs from fatal cases of SARS: PLoS Med, 2006; 3(2); e27
52. Spiegel M, Schneider K, Weber F, Interaction of severe acute respiratory syndrome-associated coronavirus with dendritic cells: J Gen Virol, 2006; 87(Pt 7); 1953-60
53. Trojanowicz B, Ulrich C, Kohler F, Monocytic angiotensin-converting enzyme 2 relates to atherosclerosis in patients with chronic kidney disease: Nephrol Dial Transplant, 2017; 32(2); 287-98
54. Abassi Z, Knaney Y, Karram T, Heyman SN, The lung macrophage in SARS-CoV-2 infection: A friend or a foe?: Front Immunol, 2020; 11; 1312
55. Jose RJ, Manuel A, COVID-19 cytokine storm: The interplay between inflammation and coagulation: Lancet Respir Med, 2020; 8(6); e46-47
56. The BASE Medicine Task Force, Facts and recommendations of SARS-CoV-2 and COVID-19: An update: Science Insights, 2020; 35(1); 194-215
57. Kremer S, Lersy F, de Sèze J, Brain MRI findings in severe COVID-19: A retrospective observational study: Radiology, 2020; 16; 202222
58. Wu K, Zou J, Chang HY, RNA-GPS predicts SARS-CoV-2 RNA localization to host mitochondria and nucleolus: Cell Syst, 2020; 11(1); 102-108.e3
59. Shenoy S, Coronavirus (Covid-19) sepsis: Revisiting mitochondrial dysfunction in pathogenesis, aging, inflammation, and mortality: Inflamm Res, 2020; 7; 1-9
60. Singh KK, Chaubey G, Chen JY, Suravajhala P, Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis: Am J Physiol Cell Physiol, 2020; 319(2); C258-67
61. Zhang AJ, Lee AC, Chu H, SARS-CoV-2 infects and damages the mature and immature olfactory sensory neurons of hamsters: Clin Infect Dis, 2020 [Online ahead of print]
62. Aghagoli G, Gallo Marin B, Katchur NJ, Neurological involvement in COVID-19 and potential mechanisms: A review: Neurocrit Care, 2020; 13; 1-10
63. Thomson BJ, Viruses and apoptosis: Int J Exp Pathol, 2001; 82(2); 65-76
64. Nainu F, Shiratsuchi A, Nakanishi Y, Induction of apoptosis and subsequent phagocytosis of virus-infected cells as an antiviral mechanism: Front Immunol, 2017; 8; 1220
65. Richard A, Tulasne D, Caspase cleavage of viral proteins, another way for viruses to make the best of apoptosis: Cell Death Dis, 2012; 3; e277
66. Shojaei S, Suresh M, Klionsky DJ, Autophagy and SARS-CoV-2 infection: Apossible smart targeting of the autophagy pathway: Virulence, 2020; 11(1); 805-10
67. Vallamkondu J, John A, Wani WY, SARS-CoV-2 pathophysiology and assessment of coronaviruses in CNS diseases with a focus on therapeutic targets: Biochim Biophys Acta Mol Basis Dis, 2020; 1866(10); 165889
68. Lukiw WJ, Pogue A, Hill JM, SARS-CoV-2 infectivity and neurological targets in the brain: Cell Mol Neurobiol, 2020; 25; 1-8
69. Trepson WL, Risk factors for Alzheimer’s disease: Science Insights, 2020; 32(2); 125-32
70. Sochocka M, Zwolińska K, Leszek J, The infectious etiology of Alzheimer’s disease: Curr Neuropharmacol, 2017; 15(7); 996-1009
71. Calderón-Garcidueñas L, Torres-Jardón R, Franco-Lira M, Environmental nanoparticles, SARS-CoV-2 brain involvement, and potential acceleration of Alzheimer’s and Parkinson’s diseases in young urbanites exposed to air pollution: J Alzheimers Dis, 2020 [Online ahead of print]
72. Chen X, Laurent S, Onur OA, A systematic review of neurological symptoms and complications of COVID-19: J Neurol, 2020 [Online ahead of print]
73. Heneka MT, Carson MJ, El Khoury J, Neuroinflammation in Alzheimer’s disease: Lancet Neurol, 2015; 14(4); 388-405
74. Guo T, Zhang D, Zeng Y, Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease: Mol Neurodegener, 2020; 15(1); 40
75. Beitz JM, Parkinson’s disease: A review: Front Biosci (Schol Ed), 2014; 6; 65-74
76. Das T, Hwang JJ, Poston KL, Episodic recognition memory and the hippocampus in Parkinson’s disease: A review: Cortex, 2019; 113; 191-209
77. Bezdicek O, Ballarini T, Buschke H, Memory impairment in Parkinson’s disease: The retrieval versus associative deficit hypothesis revisited and reconciled: Neuropsychology, 2019; 33(3); 391-405
78. Krajcovicova L, Klobusiakova P, Rektorova I, Gray matter changes in Parkinson’s and Alzheimer’s disease and relation to cognition: Curr Neurol Neurosci Rep, 2019; 19(11); 85
79. Bernaus A, Blanco S, Sevilla A, Glia crosstalk in neuroinflammatory diseases: Front Cell Neurosci, 2020; 14; 209
80. Van Bulck M, Sierra-Magro A, Alarcon-Gil J, Novel approaches for the treatment of Alzheimer’s and Parkinson’s disease: Int J Mol Sci, 2019; 20(3); 719
81. Compta Y, Parkkinen L, Kempster P, The significance of α-synuclein, amyloid-β and tau pathologies in Parkinson’s disease progression and related dementia: Neurodegener Dis, 2014; 13(2–3); 154-56
82. Sulzer D, Antonini A, Leta V, COVID-19 and possible links with Parkinson’s disease and parkinsonism: From bench to bedside: NPJ Parkinsons Dis, 2020; 6; 18
83. Ponsen MM, Stoffers D, Booij J, Idiopathic hyposmia as a preclinical sign of Parkinson’s disease: Ann Neurol, 2004; 56(2); 173-81
84. Lechien JR, Chiesa-Estomba CM, De Siati DR, Olfactory and gustatory dysfunctions as a clinical presentation of mild-to-moderate forms of the coronavirus disease (COVID-19): A multicenter European study: Eur Arch Otorhinolaryngol, 2020; 277(8); 2251-61
85. Giacomelli A, Pezzati L, Conti F, Self-reported olfactory and taste disorders in patients with severe acute respiratory coronavirus 2 infection: A cross-sectional study: Clin Infect Dis, 2020; 71(15); 889-90
86. Dell’Era V, Farri F, Garzaro G, Smell and taste disorders during COVID-19 outbreak: Cross-sectional study on 355 patients: Head Neck, 2020; 42(7); 1591-96
87. Haddadi K, Ghasemian R, Shafizad M, Basal ganglia involvement and altered mental status: A unique neurological manifestation of coronavirus disease 2019: Cureus, 2020; 12(4); e7869
88. Lassmann H, Multiple sclerosis pathology: Cold Spring Harb Perspect Med, 2018; 8(3); a028936
89. Kempuraj D, Selvakumar GP, Ahmed ME, COVID-19, mast cells, cytokine storm, psychological stress, and neuroinflammation: Neuroscientist, 2020; 26(5–6); 402-14
90. Zanin L, Saraceno G, Panciani PP, SARS-CoV-2 can induce brain and spine demyelinating lesions: Acta Neurochir (Wien), 2020; 162(7); 1491-94
91. Palao M, Fernández-Díaz E, Gracia-Gil J, Multiple sclerosis following SARS-CoV-2 infection: Mult Scler Relat Disord, 2020; 45; 102377
92. Arbour N, Day R, Newcombe J, Talbot PJ, Neuroinvasion by human respiratory coronaviruses: J Virol, 2000; 74(19); 8913-21
93. Boziki MK, Mentis AA, Shumilina M, COVID-19 immunopathology and the central nervous system: Implication for multiple sclerosis and other autoimmune diseases with associated demyelination: Brain Sci, 2020; 10(6); 345
Figures
Figure 1. Respiratory injury associated with SARS-CoV-2 infection. SARS-CoV-2 induces inflammatory changes in all 3 main locations of the respiratory system from the trachea (A), to the bronchi (B), and the alveolar sacs (C). The respiratory system will experience acute inflammation following the pro-inflammatory ‘cytokine storm,’ followed by long-term fibrotic changes. Pulmonary fibrosis is associated with the upregulation of transforming growth factor-beta (TGF-β) and chronic inflammation.Figure 2. Neuronal injury associated with SARS-CoV-2 infection. SARS-CoV-2 enters into the central nervous system (CNS) through 2 major routes: the olfactory pathway (#1 in A), and the blood–brain barrier (BBB) pathway (#2 in A). The virus can migrate into the CNS directly by endocytosis with the assistance of inflammatory cytokine-induced increased vascular permeability and indirect transfer via a ‘Trojan horse’ mechanism. (B) After binding to its membrane receptor, ACE2, SARS-CoV-2 will be engulfed into neuronal cytosol and move to connect with cytosol-located angiotensin-converting enzyme 2 (ACE2). The viral RNAs enter the mitochondria or form into autophagolysosomes to initiate autophagy and/or apoptosis. Microglia and immune cells produce pro-inflammatory cytokines, which result in further abnormalities in mitochondrial function. When the virus enters the neurons, it combines with the axonal microtubules with anterograde and retrograde spread to the synapse and enters the next level of neurons by trans-synaptic transfer and endocytosis. Both the virus and the ‘cytokine storm’ can destroy the myelin sheath of neurons, resulting in acute and chronic neuropathology. In Press
07 Mar 2024 : Clinical Research
Knowledge of and Attitudes Toward Clinical Trials: A Questionnaire-Based Study of 179 Male Third- and Fourt...Med Sci Monit In Press; DOI: 10.12659/MSM.943468
08 Mar 2024 : Animal Research
Modification of Experimental Model of Necrotizing Enterocolitis (NEC) in Rat Pups by Single Exposure to Hyp...Med Sci Monit In Press; DOI: 10.12659/MSM.943443
18 Apr 2024 : Clinical Research
Comparative Analysis of Open and Closed Sphincterotomy for the Treatment of Chronic Anal Fissure: Safety an...Med Sci Monit In Press; DOI: 10.12659/MSM.944127
08 Mar 2024 : Laboratory Research
Evaluation of Retentive Strength of 50 Endodontically-Treated Single-Rooted Mandibular Second Premolars Res...Med Sci Monit In Press; DOI: 10.12659/MSM.944110
Most Viewed Current Articles
17 Jan 2024 : Review article
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
14 Dec 2022 : Clinical Research
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387
01 Jan 2022 : Editorial
Editorial: Current Status of Oral Antiviral Drug Treatments for SARS-CoV-2 Infection in Non-Hospitalized Pa...DOI :10.12659/MSM.935952
Med Sci Monit 2022; 28:e935952