16 September 2022: Review Articles
Mechanisms of Myocardial Damage Due to Hyperlipidemia: A Review of Recent Studies
Zhiqi Zhang1EF, Hongyi Wu1F, Tao Wang2A, Yao Liu3F, Chun Meng2AG*DOI: 10.12659/MSM.937051
Med Sci Monit 2022; 28:e937051






Abstract
ABSTRACT: Myocardial injury and necrosis caused by hyperlipidemia have been investigated by several researchers. Their pathogenesis and molecular basis are different from those of the more common clinical ischemic myocardial injury. Hyperlipidemia leads to peroxide accumulation in the cardiomyocytes, causes lipid overload, decreases the antioxidant capacity of the body, and promotes the inflammatory response. Furthermore, hyperlipidemia causes changes in the structure and function of mitochondria in the cardiomyocytes, which results in their injury and necrosis. Many previous studies have shown that metabolic diseases (eg, obesity and diabetes) and chemical poisoning can lead to hyperlipidemic myocardial injury and necrosis. Moreover, it has been observed that this pathological process can be inhibited by many small molecular substances. In the clinic, myocardial damage can be prevented or reduced by lowering the levels of triglyceride and cholesterol. Myocardial damage can also be regulated via the molecular pathway of myocardial injury caused by hyperlipidemia so that the disease can be treated. The present article reviewed the recent findings reported on the mechanisms of myocardial damage due to hyperlipidemia.
Keywords: review, Hyperlipidemias, Heart Failure, Oxidative Stress, Antioxidants, Humans, Lipids, Necrosis, Peroxides, Triglycerides
Background
Hyperlipidemia refers to increased fasting triglyceride (TG) and/or total cholesterol (TC) in the blood. The condition can be subdivided into hypertriglyceridemia, hypercholesterolemia, mixed hyperlipidemia, and high levels of low-density lipoprotein (LDL). In terms of etiology, it can be divided into hyperlipidemia caused by genetic factors and non-genetic factors, also known as changeable factors and non-variable factors [1]. In recent years, researchers have also found that free fatty acids (FFAs), especially palmitic acid, play a key role in the mechanism of oxidative stress, iron death, and other areas of research [2,3].
Cardiomyocytes are damaged or even die to varying degrees depending on the disease state. Previous studies have shown that ischemic cardiomyopathy, hypertensive cardiomyopathy, and diabetic cardiomyopathy are the common causes of myocardial injury and necrosis [4,5]. Myocardial cell damage is also seen in hyperthyroidism, renal insufficiency, and viral myocarditis. The occurrence and development of the abovementioned diseases are often accompanied by abnormal lipid metabolisms, such as dyslipidemia in patients with coronary heart disease and diabetes [6]. In recent years, some researchers have stated that myocardial cell damage can occur even when hyperlipidemia has not progressed to ischemic cardiomyopathy and is not accompanied by other metabolic diseases [7–9]. Hyperlipidemia is known to be associated with coronary artery disease due to atherosclerosis and ischemic heart disease. Recently, it was revealed that hyperlipidemia is also associated with an increased risk of non-ischemic heart failure, which can, however, be reversed by controlling the lipid levels [10].
Histopathological studies have demonstrated that disordered myocardial structure, cardiomyocyte fibrosis [11], and even interstitial hemorrhage [12] can occur because of the influence of hyperlipidemia. A high-fat diet can induce cell apoptosis [13], ferroptosis, and autophagy [3,14], thus participating in various disease processes. From a biochemical perspective, a high-fat diet can induce the body’s inflammatory response [8], increase the production of reactive oxygen species (ROS) [15], downregulate the expression of nuclear factor E2-related factor 2 (Nrf2) in the antioxidant system [7], and alter mitochondrial structure and function [7,16]. However, at present, the mechanism of the hyperlipidemia-induced inflammatory response is not completely understood. Comprehensive data are not available on cardiomyocyte apoptosis induced by pure hyperlipidemia [8], autophagy, and ferroptosis; hence, further research is needed. Since many diseases and lesions involve hyperlipidemia, a systematic and in-depth understanding of the pathogenesis of this condition and the mechanism of damage to the body can aid in the diagnosis and treatment of clinical diseases.
Cardiac Lipid Accumulation
Glucose, fat, and ketone bodies are the main energy sources utilized for myocardial activity in the physiological state. The metabolic utilization of these energy substrates by myocardial tissues is significantly affected by the concentration of these substrates [17]. Under normal physiological conditions, the heart selects the corresponding substrates for energy metabolism in accordance with the amount of these energy substances, which more efficiently used the nutrients in the body and avoid the accumulation of a specific substance in the myocardium. Under certain pathological situations, such as obesity, metabolic syndrome, or ischemic cardiomyopathy, the utilization of fatty acids by the myocardial tissue decreases, leading to fat accumulation [18].
Lipid accumulation can induce pathological changes, such as inflammatory response, lipotoxicity, and cellular fibrosis, in the myocardial tissues [19]. These injuries caused by lipid accumulation in the myocardial tissues ultimately result in a decline in cardiac functions [20].
In the present review we discuss the myocardial damage induced by excessive lipid accumulation in terms of the proinflammatory response and decreased endoplasmic reticulum function and investigate the effect of high lipid concentration on the myocardial antioxidant capacity (Table 1).
Inflammatory Response Associated with Hyperlipidemia
Hyperlipidemia can alter the levels of inflammatory factors in the body. Some studies have shown that in the animal model of hypercholesterolemia, the level of plasma Cytokine-induced neutrophil chemokine-1 (CINC-1) increases, and the level of anti-inflammatory chemokine adiponectin decreases. On the contrary, the levels of inflammatory factors, such as CINC-1, interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α), in the myocardium decrease significantly, and the level of the anti-inflammatory factor adiponectin increases significantly [8]. The decrease of inflammatory factors and the increase of anti-inflammatory factors in the myocardium may be a self-protective mechanism of the cardiomyocytes in the early stages of hypercholesterolemia. To verify this conjecture, long-term animal experiments should be conducted on hypercholesterolemia to explore the changes in inflammatory and anti-inflammatory factors in cardiomyocytes after prolonged hypercholesterolemia.
Peroxisome proliferator-activated receptor (PPAR) is mainly distributed in the adipocytes. Research advancements have provided more information on the functions and distribution of PPARs. Researchers have identified that PPAR and nuclear factor κB (NF-κB) mutually inhibit each other, thus playing an important role in anti-inflammation [29]. The level of LDL in the serum increases and the level of oxLDL increases accordingly, which can activate NF-κB [30]. After the activation of NF-κB, on the one hand, it promotes the
Increased levels of TG in the blood also stimulate the body’s inflammatory response. After phagocytosis of lipids, foam cells formed by the macrophages participate in the inflammatory process of many diseases [40]. While TG promotes the activation of macrophages, excessive FFAs can induce inflammation and cause damage to multiple organs [41]. On the contrary, when the synthesis of TG is inhibited and the level of FFAs in the blood decreases, the phagocytosis of macrophages and the function of secreting inflammatory factors (IL-1 β, IL-6, and PGE2) is also inhibited [42] (see Figure 1 for the specific proinflammatory mechanisms of TC and TG).
Hyperlipidemia Leads to Oxidative Stress
It has been documented that the level of serum malondialdehyde increases in the animal model of hyperlipidemia, which suggests that hyperlipidemia could cause lipid peroxidation
ROS are usually produced during the oxidative phosphorylation of lipids by the mitochondria. The massive accumulation of ROS is often inseparable from the excessive activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) [47]. With the increase in the OxLDL level, ROS accumulation is induced by the oxLDL/receptor for advanced glycation end-product (RAGE)/NADPH oxidase NOX pathway [48]. It has been observed that oxLDL can also promote ROS production via lectin-like oxidized low-density lipoprotein recaptor-1 (LOX-1) [49] and CD36 [50] pathways, all of which are a part of the NOX family. The accumulation of ROS triggered by high fat levels causes damage to the cells. In this process, the body’s response is often to produce antioxidants and antioxidative damage, but in cells with excessive damage, apoptosis or necrosis is induced [37]. NF-κB not only regulates inflammation but also exerts an antioxidant effect, and antioxidant enzymes, such as manganese superoxide dismutase, can be used as targets of NF-κB [43]. In the course of the disease, ROS inhibits the antioxidant pathway of NF-κB and also enhances the proinflammatory pathway of NF-κB [51].
Furthermore, several studies have shown that the levels of oxidizing substances, such as oxLDL and ROS, promote the activation and expression of Nrf2 and increase the level of downstream antioxidant products [52]. Nrf2 is an important internal factor in the body, which plays a key role in antioxidation and antistress and offers protection against a variety of related diseases [53]. Free Nrf2 enters the nucleus and binds to Maf protein, which then activates the cis-acting element ARE and induces the production of downstream substances. [54]. Its downstream products include heme oxidase-1 (HO-1), glutathione, quinone dehydrogenase 1, thioredoxin, and other important antioxidant proteins [55–57]. Experimental results have established that the myocardial damage caused by hyperlipidemia can be effectively reduced by activating the Nrf2 antioxidant system [7]. However, in some animal experiments it has been observed that in the hyperlipidemic disease model established by a high-fat diet combined with Poloxamer 407 (P407) injection, the levels of antioxidants, such as Nrf2 and HO-1, were significantly lower than those in the control group, and the peroxidation reaction was increased. On the contrary, in the experimental group in which Nrf2 was activated, the oxidative damage of the target organs decreased significantly [58–60]. Unfortunately, the mechanism of hyperlipidemia inhibiting Nrf2 expression has not been further explored. We suspect that this may be due to oxidative damage causing excessive damage to the cells, which cannot effectively protect themselves against oxidation. However, this conjecture needs to be verified with more experiments, and its specific molecular mechanism and pathway need to be clarified. Hence, it is evident that a variety of cell injury and necrosis pathways are involved in oxidative stress. The study of antioxidation is an important link in the treatment of hyperlipidemic myocardial injury (see Figure 1 for the pathway of oxidative stress damage caused by high fat).
Changes in the Endoplasmic Reticulum and Mitochondria Caused by Hyperlipidemia
Researchers have stated that hyperlipidemia can damage mitochondrial function [61]. High fatty acid levels in the blood can promote the acetylation of mitochondrial fission protein power-related protein 1 (Drp1). In the intracellular environment of Drp1 acetylation, mitochondria exhibit structural ectopia and decreased function, which leads to myocardial dysfunction [62]. High fat can also affect the endoplasmic reticulum (ER). The accumulation of FFAs in the blood causes stress damage to the ER, which results in a decline in the metabolic capacity of the body and then affects the function of various organs and tissues [63]. Moreover, the accumulation of FFAs leads to the misfolding of ER molecular chaperones, which damages the structure and function of the ER [64]. High FFA levels increase ROS
FFAs are oxidized in the mitochondria to produce adenosine triphosphate, which provides energy for the body. Mitochondrial dysfunction can reduce the function of cardiomyocytes and eventually lead to a decline in cardiac function [66]. The metabolism of FFAs in the mitochondria is regulated by PPARs that promote this metabolic process [67,68]. The expression of PPARs is inhibited by NF-κB. When the expression of NF-κB increases, the expression of PPARs is inhibited, thus affecting the metabolism of FFAs and lowering the myocardial energy supply and cardiac function [69]. When the level of FFAs in the blood increases, their metabolism does not correspondingly increase, but activates NF-κB in the tissues [70,71], resulting in a decrease in the metabolic function of mitochondria toward FFAs. To change this pathological process, in addition to the various hypolipidemic schemes mentioned above, we can also promote mitochondrial metabolism and increase the myocardial energy supply by inhibiting NF-κB and activating PPARs, which improves cardiac functions [72].
As mentioned above, oxLDL also inhibits the function of PPARs by activating NF-κB, which undoubtedly affects the metabolic function of mitochondria toward FFAs. These findings suggest that NF-κB may be a key target to improve the mitochondrial dysfunction caused by hyperlipidemia. It is known that cholesterol in the body is transported to the liver for a series of metabolic processes. Cholesterol entering the liver is converted not only to bile acids but also to steroids in the mitochondria [73]. oxLDL clearly promotes the production of ROS, and excessive ROS leads to ER and mitochondrial stress, which in turn trigger the production of more ROS, thus affecting the metabolic function of the mitochondria [74]. Stress injury in the ER undoubtedly leads to further pathological damage to tissues and organs [75]. In severe cases, it causes a decline in the function of various organs, including the heart. In addition to the ROS pathway, oxLDL itself can induce ER stress and trigger a series of subsequent reactions, causing protein folding disorder and apoptosis. The specific pathway of ER stress leading to apoptosis is relatively complex and is not described in detail here. Hence, please refer to the relevant literature [76] (see Figure 2 for the pathway of ER and mitochondrial stress caused by hyperlipidemia).
Notably, fatty acid metabolism is cross-linked with inflammatory reaction and oxidative stress via NF-κB, ROS, and PPARs. A series of pathological changes caused by hyperlipidemia are not independent of each other; rather, they have certain interactions.
LIPID-LOWERING AND ANTI-INFLAMMATORY THERAPY:
In a clinical setting, the most commonly employed and effective approach is to reduce the LDL level and simultaneously create a corresponding anti-inflammatory effect. The primary ways to reduce TC are to reduce its absorption and synthesis and promote its degradation. For example, statins can clearly reduce the level of TC and inhibit inflammation. Moreover, the TC transporter NPC1-like1 inhibitor ezetimibe and proprotein-converting enzyme subtilisin 9 (PCSK9) inhibitor are recommended to treat high cholesterol levels [77]. As per the reports of large clinical double-blinded controlled trials, PCSK9 inhibitors significantly reduced the LDL levels of the patients [78]. Several PCSK9 inhibitors have been applied clinically with good outcomes in controlling the blood lipid levels [79]. However, the currently approved PCSK9 inhibitors can only be used as injectables and their price is higher than that of other lipid-lowering drugs. Nevertheless, the good news is that oral PCSK9 inhibitors are currently being developed and tested, which would undoubtedly and significantly improve patient compliance with the prescribed medication [80,81]. As per the TC management guidelines, the roles of lifestyle, niacin derivatives, and cholesteryl ester transfer protein inhibitors in the control of TC have been emphasized [82].
Presently, PPARα agonist fibrate lipid-lowering drugs are widely applied clinically, which not only regulate the metabolic levels of TGs and lipoproteins but also alleviate the inflammatory injury caused by high FFAs [41]. Supplementation with unsaturated fatty acids is an option to reduce triglycerides. Omega-3 is clinically used to lower triglycerides and combat the inflammatory response caused by hyperlipidemia [83]. Similarly, the levels of TGs and FFAs in the body can be reduced through lifestyle interventions or supplementation with nicotinic acid derivatives.
ANTIOXIDANT THERAPY:
In addition, some phytochemicals, such as resveratrol [84,85] and Herba houttuyniae extract [7], have demonstrated their antioxidant capacity in experiments and displayed a protecting role in the myocardium under high-fat conditions. Another plant extract, coenzyme Q10, is an important dietary supplement with cardioprotective effects [86]. Coenzyme Q10 is involved in the oxidative phosphorylation process of the mitochondria, which is an important antioxidant substance with a clear antioxidant role in the disease process [87,88]. Moreover, coenzyme Q10 plays a role in counteracting inflammation [89] and lowering cholesterol [90]. More relevant studies are needed on such phytochemicals before they can be widely applied in clinical practice.
IMPROVEMENT OF MYOCARDIAL METABOLISM:
Trimetazidine, as classical prazosin, plays an important role in mediating myocardial metabolism. It can reduce the uptake of FFAs [91] and strengthen the utilization of glucose by the mitochondria [92], thereby playing a role in protecting the myocardium [93]. As a drug of the same class, ranolazine is effective in improving the myocardial metabolism [94] and in regulating the systemic metabolism in some experiments [95]. Increasing research has demonstrated that ranolazine has a more mechanistic involvement in its antianginal effect [96], but it is not described in detail here.
In addition, some drugs that regulate systemic energy metabolism have also achieved improvement in myocardial metabolism. Piercillin [97] and etomox [98] enhanced glucose utilization in cardiomyocytes by inhibiting carnitine palmitoyl transferase 1, thereby counteracting the adverse effects of lipid accumulation on the myocardial metabolism. Pyruvate dehydrogenase kinase inhibitors have also been demonstrated to enhance myocardial metabolism in the past [99,100]. However, more research is needed in these areas before their clinical application.
Discussion
The heart acts as an important circulatory organ that can use carbohydrates, fats, and ketones as energy sources [17]. It consumes excessive energy at all times during physiological activities; therefore, the metabolic functions of cardiomyocytes are extremely important for the heart. Excessive lipids can affect heart metabolism and even decrease cardiac functions [10,20]. However, there is no clear definition and diagnostic criteria for “lipotoxic cardiomyopathy” [101]. Fortunately, further studies on ischemic cardiomyopathy, diabetic cardiomyopathy, and heart failure have emphasized the need for research on myocardial metabolic functions. In the future, it will be critical to comprehend the mechanism and pathological changes of hyperlipidemic myocardial injury and provide a theoretical basis for the diagnosis and treatment of the hyperlipidemic myocardial injury.
Conclusions
Hyperlipidemia, as one of the main components of metabolic syndrome, can cause damage to multiple organs [102]. The damage caused by hyperlipidemia to cardiomyocytes and other tissues and organs is mainly caused by an inflammatory reaction, structural and functional changes in the ER and mitochondria, and lipid peroxidation [66]. Presently, considerable achievements have been recorded in controlling blood lipid levels through clinical work [1,102,103]. With the in-depth study of the mechanism of hyperlipidemic myocardial injury, newer methods can be applied to treat myocardial injury and alleviate the cardiac function decline caused by hyperlipidemia, thereby benefiting patients.
Figures
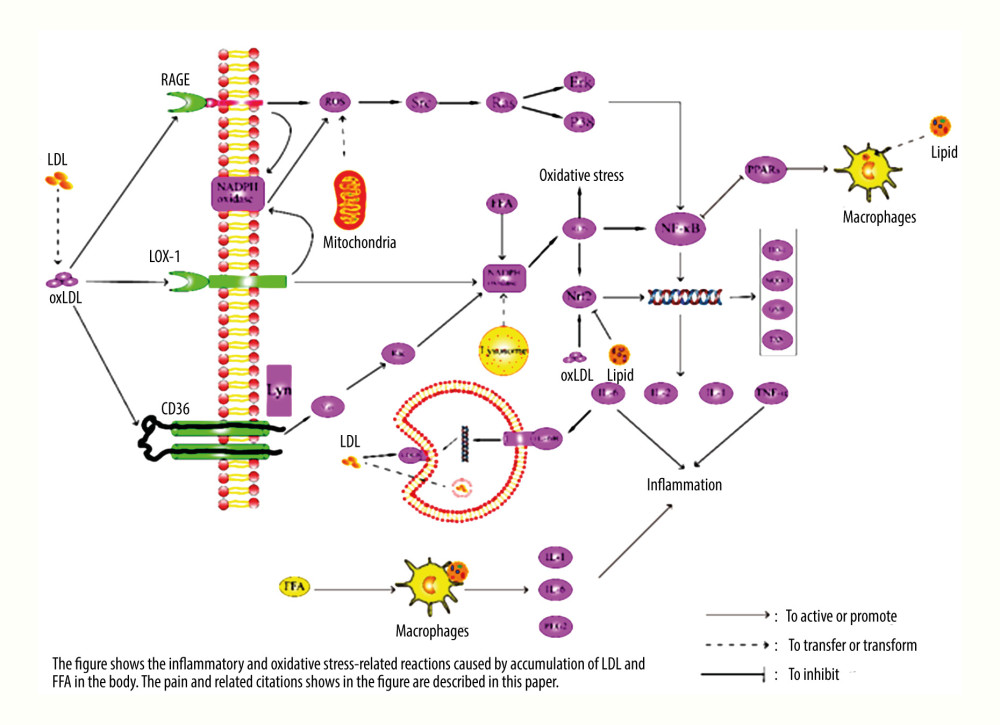 Figure 1. Pathway diagram of hyperlipidemia leading to inflammatory response and oxidative stress. LDL – low density lipoprotein; FFA – free fatty acid; ROS – reactive oxygen species; SRC – tyrosine-protein kinase Src; RAS – GTPase HRas; ERK – mitogen-activated protein kinase; P38 – P38 mitogen-activated protein kinase; VAV – guanine nucleotide exchange factor VAV; Rac – Ras-related C3 botulinum toxin substrate; Lyn – tyrosine-proteon kinase Lyn; TXN – thierodexin; GSH – glutathione; oxLDL – oxidized low density lipoprotein; LOX-1 – lectin-like oxidized low-density lipoprotein receptor-1; RAGE – advanced glycation end product receptor; CD36 – CD36 antigen; LDLR – LDL receptor; PPARs – peroxisome proliferators-activated receptors; HO-1 – heme oxygenate-1; NQO-1 – quinine oxidoreductase 1; NF-κB – nuclear factor-κB; Nrf2 – nuclear factor erythroid 2-related factor 2; TNF-α – tumour necrosis factor alpha; IL – interleukin.
Figure 1. Pathway diagram of hyperlipidemia leading to inflammatory response and oxidative stress. LDL – low density lipoprotein; FFA – free fatty acid; ROS – reactive oxygen species; SRC – tyrosine-protein kinase Src; RAS – GTPase HRas; ERK – mitogen-activated protein kinase; P38 – P38 mitogen-activated protein kinase; VAV – guanine nucleotide exchange factor VAV; Rac – Ras-related C3 botulinum toxin substrate; Lyn – tyrosine-proteon kinase Lyn; TXN – thierodexin; GSH – glutathione; oxLDL – oxidized low density lipoprotein; LOX-1 – lectin-like oxidized low-density lipoprotein receptor-1; RAGE – advanced glycation end product receptor; CD36 – CD36 antigen; LDLR – LDL receptor; PPARs – peroxisome proliferators-activated receptors; HO-1 – heme oxygenate-1; NQO-1 – quinine oxidoreductase 1; NF-κB – nuclear factor-κB; Nrf2 – nuclear factor erythroid 2-related factor 2; TNF-α – tumour necrosis factor alpha; IL – interleukin. 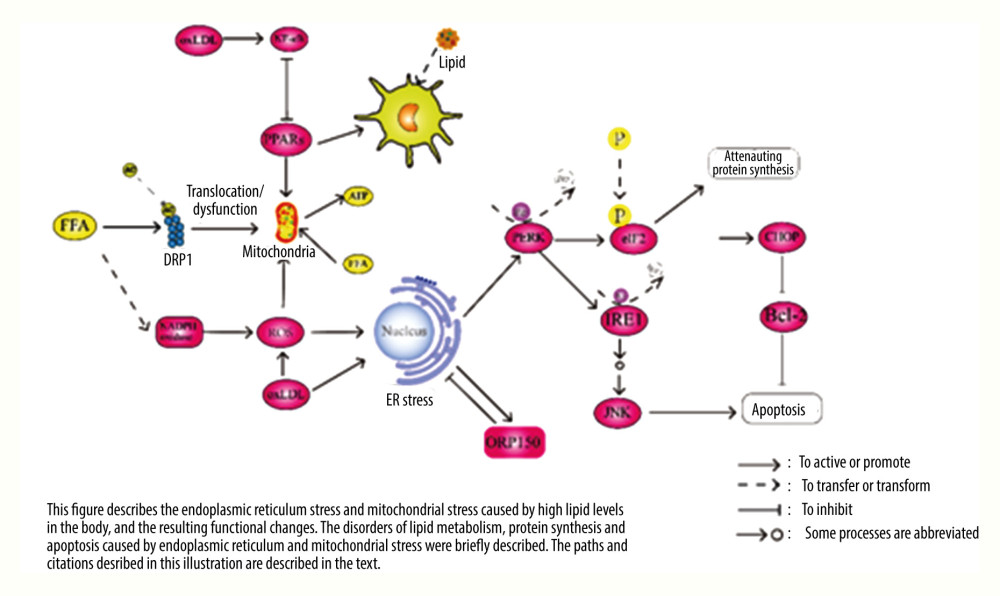 Figure 2. Pathway diagram of myocardial endoplasmic reticulum and mitochondria damage caused by hyperlipidemia. FFA – free fatty acid; ROS – reactive oxygen species; JNK – c-Jun N-terminal kinase; ATP – adenosine triphosphate; ER – endoplasmic reticulum; DRP1 – dynamin-related protein 1; AC – acetyl group; Bip – binding immunoglobulin protein; P – phosphoryl group; oxLDL – oxidized low density lipoprotein; PERK – double-stranded RNA-dependent protein kinase-like ER kinase; eIF2 – eukaryotic initation factor 2; IRE1 – inositol-requiring enzyme 1; Bcl-2 – B-cell lymphoma-2; CHOP – the C/EBP homologous protein; PPARs – peroxisome proliferators-activated receptors; NF-κB – nuclear factor-κB.
Figure 2. Pathway diagram of myocardial endoplasmic reticulum and mitochondria damage caused by hyperlipidemia. FFA – free fatty acid; ROS – reactive oxygen species; JNK – c-Jun N-terminal kinase; ATP – adenosine triphosphate; ER – endoplasmic reticulum; DRP1 – dynamin-related protein 1; AC – acetyl group; Bip – binding immunoglobulin protein; P – phosphoryl group; oxLDL – oxidized low density lipoprotein; PERK – double-stranded RNA-dependent protein kinase-like ER kinase; eIF2 – eukaryotic initation factor 2; IRE1 – inositol-requiring enzyme 1; Bcl-2 – B-cell lymphoma-2; CHOP – the C/EBP homologous protein; PPARs – peroxisome proliferators-activated receptors; NF-κB – nuclear factor-κB. 
References
1. Karr S, Epidemiology and management of hyperlipidemia: Am J Manag Care, 2017; 23(9 Suppl); S139-48
2. Henderson GC, Plasma free fatty acid concentration as a modifiable risk factor for metabolic disease: Nutrients, 2021; 13(8); 2590
3. Ma W-Q, Sun X-J, Zhu Y, Liu N-F, Metformin attenuates hyperlipidaemia-associated vascular calcification through anti-ferroptotic effects: Free Radic Biol Med, 2021; 165; 229-42
4. Crisafulli A, Pagliaro P, Roberto S, Diabetic cardiomyopathy and ischemic heart disease: Prevention and therapy by exercise and conditioning: Int J Mol Sci, 2020; 21(8); 2896
5. Mouton AJ, Li X, Hall ME, Hall JE, Obesity, hypertension, and cardiac dysfunction: Novel roles of immunometabolism in macrophage activation and inflammation: Circ Res, 2020; 126(6); 789-806
6. Powell-Wiley TM, Poirier P, Burke LE, Obesity and cardiovascular disease: A scientific statement from the American Heart Association: Circulation, 2021; 143(21); e984-e1010
7. Cao K, Lv W, Liu X, Herba houttuyniae extract benefits hyperlipidemic mice via activation of the AMPK/PGC-1α/Nrf2 cascade: Nutrients, 2020; 12(1); 164
8. Han Q, Yeung SC, Ip MSM, Mak JCW, Dysregulation of cardiac lipid parameters in high-fat high-cholesterol diet-induced rat model: Lipids Health Dis, 2018; 17(1); 255
9. Xu J, Zhu L, Liu H, Thymoquinone reduces cardiac damage caused by hypercholesterolemia in apolipoprotein E-deficient mice: Lipids Health Dis, 2018; 17(1); 173
10. Yao YS, Di Li T, Zeng ZH, Mechanisms underlying direct actions of hyperlipidemia on myocardium: An updated review: Lipids Health Dis, 2020; 19(1); 23
11. Chen X, Yu W, Li W, An anti-inflammatory chalcone derivative prevents heart and kidney from hyperlipidemia-induced injuries by attenuating inflammation: Toxicol Appl Pharmacol, 2018; 338; 43-53
12. AlSaad AMS, Alasmari F, Abuohashish HM, Renin angiotensin system blockage by losartan neutralize hypercholesterolemia-induced inflammatory and oxidative injuries: Redox Rep, 2020; 25(1); 51-58
13. Li K, Deng Y, Deng G, High cholesterol induces apoptosis and autophagy through the ROS-activated AKT/FOXO1 pathway in tendon-derived stem cells: Stem Cell Res Ther, 2020; 11(1); 131
14. Xie Y, Li J, Kang R, Tang D, Interplay between lipid metabolism and autophagy: Front Cell Dev Biol, 2020; 8; 431
15. Csonka C, Sárközy M, Pipicz M, Modulation of hypercholesterolemia-induced oxidative/nitrative stress in the heart: Oxid Med Cell Longev, 2016; 2016; 3863726
16. Chtourou Y, Slima AB, Makni M, Naringenin protects cardiac hypercholesterolemia-induced oxidative stress and subsequent necroptosis in rats: Pharmacol Rep, 2015; 67(6); 1090-97
17. Zuurbier CJ, Bertrand L, Beauloye CR, Cardiac metabolism as a driver and therapeutic target of myocardial infarction: J Cell Mol Med, 2020; 24(11); 5937-54
18. Gupta R, Ranchal P, Mahajan S, Lipid inclusions in cardiac myocytes – a rare case of cardiolipotoxicity: Future Cardiol, 2021; 17(2); 293-99
19. Hiruma S, Shigiyama F, Hisatake S, A prospective randomized study comparing effects of empagliflozin to sitagliptin on cardiac fat accumulation, cardiac function, and cardiac metabolism in patients with early-stage type 2 diabetes: the ASSET study: Cardiovasc Diabetol, 2021; 20(1); 32
20. Goldberg IJ, Trent CM, Schulze PC, Lipid metabolism and toxicity in the heart: Cell Metab, 2012; 15(6); 805-12
21. Lubrano V, Gabriele M, Puntoni MR, Relationship among IL-6, LDL cholesterol and lipid peroxidation: Cell Mol Biol Lett, 2015; 20(2); 310-22
22. Nakagomi A, Seino Y, Noma S, Relationships between the serum cholesterol levels, production of monocyte proinflammatory cytokines and long-term prognosis in patients with chronic heart failure: Intern Med, 2014; 53(21); 2415-24
23. Addisu A, Gower WR, Serrano M, Heart failure mice exhibit decreased gastric emptying and intestinal absorption: Exp Biol Med (Maywood), 2011; 236(12); 1454-60
24. Gierens H, Nauck M, Roth M, Interleukin-6 stimulates LDL receptor gene expression via activation of sterol-responsive and Sp1 binding elements: Arterioscler Thromb Vasc Biol, 2000; 20(7); 1777-83
25. Ye Q, Chen Y, Lei H, Inflammatory stress increases unmodified LDL uptake via LDL receptor: An alternative pathway for macrophage foam-cell formation: Inflamm Res, 2009; 58(11); 809-18
26. Catapano AL, Pirillo A, Norata GD, Vascular inflammation and low-density lipoproteins: Is cholesterol the link? A lesson from the clinical trials: Br J Pharmacol, 2017; 174(22); 3973-85
27. Kawashiri S, Kawakami A, Yamasaki S, Effects of the anti-interleukin-6 receptor antibody, tocilizumab, on serum lipid levels in patients with rheumatoid arthritis: Rheumatol Int, 2011; 31(4); 451-56
28. Strang AC, Bisoendial RJ, Kootte RS, Pro-atherogenic lipid changes and decreased hepatic LDL receptor expression by tocilizumab in rheumatoid arthritis: Atherosclerosis, 2013; 229(1); 174-81
29. Duan SZ, Usher MG, Mortensen RM, Peroxisome proliferator-activated receptor-gamma-mediated effects in the vasculature: Circ Res, 2008; 102(3); 283-94
30. Kattoor AJ, Kanuri SH, Mehta JL, Role of Ox-LDL and LOX-1 an Atherogenesis: Curr Med Chem, 2019; 26(9); 1693-700
31. Heida A, Gruben N, Catrysse L, The hepatocyte IKK: NF-κB axis promotes liver steatosis by stimulating de novo lipogenesis and cholesterol synthesis: Mol Metab, 2021; 54; 101349
32. Mullur R, Liu Y-Y, Brent GA, Thyroid hormone regulation of metabolism: Physiol Rev, 2014; 94(2); 355-82
33. Chambers KF, Day PE, Aboufarrag HT, Kroon PA, Polyphenol effects on cholesterol metabolism via bile acid biosynthesis, CYP7A1: A review: Nutrients, 2019; 11(11); 2588
34. König B, Koch A, Spielmann J, Hilgenfeld C, Activation of PPARalpha lowers synthesis and concentration of cholesterol by reduction of nuclear SREBP-2: Biochem Pharmacol, 2007; 73(4); 574-85
35. Kliewer SA, Sundseth SS, Jones SA, Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma: Proc Natl Acad Sci USA, 1997; 94(9); 4318-23
36. Christofides A, Konstantinidou E, Jani C, Boussiotis VA, The role of peroxisome proliferator-activated receptors (PPAR) in immune responses: Metabolism, 2021; 114; 154338
37. Moore KJ, Rosen ED, Fitzgerald ML, The role of PPAR-gamma in macrophage differentiation and cholesterol uptake: Nat Med, 2001; 7(1); 41-47
38. Bojic LA, Sawyez CG, Telford DE, Activation of peroxisome proliferator-activated receptor δ inhibits human macrophage foam cell formation and the inflammatory response induced by very low-density lipoprotein: Arterioscler Thromb Vasc Biol, 2012; 32(12); 2919-28
39. Mirza AZ, Althagafi II, Shamshad H, Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications: Eur J Med Chem, 2019; 166; 502-13
40. Guerrini V, Gennaro ML, Foam cells: One size doesn’t fit all: Trends Immunol, 2019; 40(12); 1163-79
41. Tao H, Yancey PG, Blakemore JL, Macrophage SR-BI modulates autophagy via VPS34 complex and PPARα transcription of Tfeb in atherosclerosis: J Clin Invest, 2021; 131(7); e94229
42. Castoldi A, Monteiro LB, van Teijlingen Bakker N, Triacylglycerol synthesis enhances macrophage inflammatory function: Nat Commun, 2020; 11(1); 4107
43. Li Q, Sun Y, Liu B, ACT001 modulates the NF-κB/MnSOD/ROS axis by targeting IKKβ to inhibit glioblastoma cell growth: J Mol Med (Berl), 2020; 98(2); 263-77
44. Yang S, Lian G, ROS and diseases: Role in metabolism and energy supply: Mol Cell Biochem, 2020; 467(1–2); 1-12
45. Gai Z, Wang T, Visentin M, Kullak-Ublick GA, Lipid accumulation and chronic kidney disease: Nutrients, 2019; 11(4); 722
46. Pinti MV, Fink GK, Hathaway QA, Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis: Am J Physiol Endocrinol Metab, 2019; 316(2); E268-85
47. Bedard K, Krause K-H, The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology: Physiol Rev, 2007; 87(1); 245-313
48. Eun SY, Park SW, Lee JH, P2Y(2)R activation by nucleotides released from oxLDL-treated endothelial cells (ECs) mediates the interaction between ECs and immune cells through RAGE expression and reactive oxygen species production: Free Radic Biol Med, 2014; 69; 157-66
49. Kattoor AJ, Goel A, Mehta JL, LOX-1: Regulation, signaling and its role in atherosclerosis: Antioxidants (Basel), 2019; 8(7); 218
50. Cho K, Choi SH, ASK1 mediates apoptosis and autophagy during oxLDL-CD36 signaling in senescent endothelial cells: Oxid Med Cell Longev, 2019; 2019; 2840437
51. Morgan MJ, Liu Z, Crosstalk of reactive oxygen species and NF-κB signaling: Cell Res, 2011; 21(1); 103-15
52. Alonso-Piñeiro JA, Gonzalez-Rovira A, Sánchez-Gomar I, Nrf2 and heme oxygenase-1 involvement in atherosclerosis related oxidative stress: Antioxidants (Basel), 2021; 10(9); 1463
53. Yamamoto M, Kensler TW, Motohashi H, The KEAP1-NRF2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis: Physiol Rev, 2018; 98(3); 1169-203
54. Bellezza I, Giambanco I, Minelli A, Donato R, Nrf2-Keap1 signaling in oxidative and reductive stress: Biochim Biophys Acta Mol Cell Res, 2018; 1865(5); 721-33
55. Tonelli C, Chio IIC, Tuveson DA, Transcriptional regulation by Nrf2: Antioxid Redox Signal, 2018; 29(17); 1727-45
56. Loboda A, Damulewicz M, Pyza E, Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism: Cell Mol Life Sci, 2016; 73(17); 3221-47
57. Furfaro AL, Traverso N, Domenicotti C, The Nrf2/HO-1 axis in cancer cell growth and chemoresistance: Oxid Med Cell Longev, 2016; 2016; 1958174
58. Raish M, Ahmad A, Bin Jardan YA, Sinapic acid ameliorates cardiac dysfunction and cardiomyopathy by modulating NF-κB and Nrf2/HO-1 signaling pathways in streptozocin induced diabetic rats: Biomed Pharmacother, 2022; 145; 112412
59. Shen B, Zhao C, Wang Y, Aucubin inhibited lipid accumulation and oxidative stress via Nrf2/HO-1 and AMPK signalling pathways: J Cell Mol Med, 2019; 23(6); 4063-75
60. Uppin V, Acharya P, Bettadaiah Bheemanakere K, Talahalli RR, Hyperlipidemia downregulate brain antioxidant defense enzymes and neurotrophins in rats: Assessment of the modulatory potential of EPA+DHA and Zerumbone: Mol Nutr Food Res, 2020; 64(20); e2000381
61. Engin AB, What is lipotoxicity?: Adv Exp Med Biol, 2017; 960; 197-220
62. Hu Q, Zhang H, Gutiérrez Cortés N, Increased Drp1 acetylation by lipid overload induces cardiomyocyte death and heart dysfunction: Circ Res, 2020; 126(4); 456-70
63. Zingg J-M, Hasan ST, Meydani M, Molecular mechanisms of hypolipidemic effects of curcumin: Biofactors, 2013; 39(1); 101-21
64. Karaskov E, Scott C, Zhang L, Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis: Endocrinology, 2006; 147(7); 3398-407
65. Brookheart RT, Michel CI, Listenberger LL, The non-coding RNA gadd7 is a regulator of lipid-induced oxidative and endoplasmic reticulum stress: J Biol Chem, 2009; 284(12); 7446-54
66. Zhang X, Li M, Wang H, Astragaloside IV alleviates the myocardial damage induced by lipopolysaccharide via the toll-like receptor 4 (TLR4)/nuclear factor kappa B (NF-κB)/proliferator-activated receptor α (PPARα) signaling pathway: Med Sci Monit, 2019; 25; 7158-68
67. Suzuki M, Nakamura F, Taguchi E, 4′,6-Dimethoxyisoflavone-7-O-β-D-glucopyranoside (wistin) is a peroxisome proliferator-activated receptor α (PPARα) agonist in mouse hepatocytes: Mol Cell Biochem, 2018; 446(1–2); 35-41
68. Du H, Li C, Wang Z, Effects of Danhong injection on dyslipidemia and cholesterol metabolism in high-fat diets fed rats: J Ethnopharmacol, 2021; 274; 114058
69. Chang X, Zhang K, Zhou R, Cardioprotective effects of salidroside on myocardial ischemia-reperfusion injury in coronary artery occlusion-induced rats and Langendorff-perfused rat hearts: Int J Cardiol, 2016; 215; 532-44
70. van Beek M, Oravecz-Wilson KI, Delekta PC, Bcl10 links saturated fat overnutrition with hepatocellular NF-κB activation and insulin resistance: Cell Rep, 2012; 1(5); 444-52
71. Balzan S, Lubrano V, LOX-1 receptor: A potential link in atherosclerosis and cancer: Life Sci, 2018; 198; 79-86
72. Ji Y-Y, Liu J-T, Liu N, PPARalpha activator fenofibrate modulates angiotensin II-induced inflammatory responses in vascular smooth muscle cells via the TLR4-dependent signaling pathway: Biochem Pharmacol, 2009; 78(9); 1186-97
73. Musso G, Gambino R, Cassader M, Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis: Prog Lipid Res, 2013; 52(1); 175-91
74. Cao SS, Kaufman RJ, Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease: Antioxid Redox Signal, 2014; 21(3); 396-413
75. Zuo S, Kong D, Wang C, CRTH2 promotes endoplasmic reticulum stress-induced cardiomyocyte apoptosis through m-calpain: EMBO Mol Med, 2018; 10(3); e8237
76. Sanson M, Augé N, Vindis C, Oxidized low-density lipoproteins trigger endoplasmic reticulum stress in vascular cells: Prevention by oxygen-regulated protein 150 expression: Circ Res, 2009; 104(3); 328-36
77. Wilson PWF, Polonsky TS, Miedema MD, Systematic Rreview for the 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines: Circulation, 2019; 139(25); e1144-e61
78. Sabatine MS, Giugliano RP, Keech AC, Evolocumab and clinical outcomes in patients with cardiovascular disease: N Engl J Med, 2017; 376(18); 1713-22
79. Pasta A, Cremonini AL, Pisciotta L, PCSK9 inhibitors for treating hypercholesterolemia: Expert Opin Pharmacother, 2020; 21(3); 353-63
80. Salaheldin TA, Godugu K, Bharali DJ, Novel oral nano-hepatic targeted anti-PCSK9 in hypercholesterolemia: Nanomedicine, 2022; 40; 102480
81. Gennemark P, Walter K, Clemmensen N, An oral antisense oligonucleotide for PCSK9 inhibition: Sci Transl Med, 2021; 13(593); eabe9117
82. Grundy SM, Stone NJ, Bailey AL, 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: Executive summary: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines: Circulation, 2019; 139(25); e1046-e81
83. Ruscica M, Sirtori CR, Carugo S, Omega-3 and cardiovascular prevention - Is this still a choice?: Pharmacol Res, 2022; 182; 106342
84. Penumathsa SV, Koneru S, Samuel SM, Strategic targets to induce neovascularization by resveratrol in hypercholesterolemic rat myocardium: role of caveolin-1, endothelial nitric oxide synthase, hemeoxygenase-1, and vascular endothelial growth factor: Free Radic Biol Med, 2008; 45(7); 1027-34
85. Rašković A, Ćućuz V, Torović L, Resveratrol supplementation improves metabolic control in rats with induced hyperlipidemia and type 2 diabetes: Saudi Pharm J, 2019; 27(7); 1036-43
86. Arenas-Jal M, Suñé-Negre JM, García-Montoya E, Coenzyme Q10 supplementation: Efficacy, safety, and formulation challenges: Compr Rev Food Sci Food Saf, 2020; 19(2); 574-94
87. Garrido-Maraver J, Cordero MD, Oropesa-Avila M, Clinical applications of coenzyme Q10: Front Biosci (Landmark Ed), 2014; 19(4); 619-33
88. Hargreaves I, Heaton RA, Mantle D, Disorders of human coenzyme Q10 metabolism: An overview: Int J Mol Sci, 2020; 21(18); 6695
89. Schmelzer C, Lindner I, Rimbach G, Functions of coenzyme Q10 in inflammation and gene expression: Biofactors, 2008; 32(1–4); 179-83
90. Zozina VI, Covantev S, Goroshko OA, Coenzyme Q10 in cardiovascular and metabolic diseases: Current state of the problem: Curr Cardiol Rev, 2018; 14(3); 164-74
91. Mansur AJ, Cardiac effects of trimetazidine in diabetic rats: Arq Bras Cardiol, 2019; 112(2); 179
92. Marzilli M, Vinereanu D, Lopaschuk G, Trimetazidine in cardiovascular medicine: Int J Cardiol, 2019; 293; 39-44
93. Zhirov IV, Osmolovskaya YF, Tereshchenko SN, Trimetazidine in the treatment of chronic heart failure: Kardiologiia, 2016; 56(1); 79-85
94. McCormack JG, Barr RL, Wolff AA, Lopaschuk GD, Ranolazine stimulates glucose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts: Circulation, 1996; 93(1); 135-42
95. Cassano V, Leo A, Tallarico M, Metabolic and cognitive effects of ranolazine in type 2 diabetes mellitus: Data from an in vivo model: Nutrients, 2020; 12(2); 382
96. Kaplan A, Amin G, Abidi E, Role of ranolazine in heart failure: From cellular to clinic perspective: Eur J Pharmacol, 2022; 919; 174787
97. Chong C-R, Sallustio B, Horowitz JD, Drugs that affect cardiac metabolism: Focus on perhexiline: Cardiovasc Drugs Ther, 2016; 30(4); 399-405
98. Lundsgaard A-M, Fritzen AM, Nicolaisen TS, Glucometabolic consequences of acute and prolonged inhibition of fatty acid oxidation: J Lipid Res, 2020; 61(1); 10-19
99. Le Page LM, Rider OJ, Lewis AJ, Increasing pyruvate dehydrogenase flux as a treatment for diabetic cardiomyopathy: A combined 13C hyperpolarized magnetic resonance and echocardiography study: Diabetes, 2015; 64(8); 2735-43
100. Wu C-Y, Satapati S, Gui W, A novel inhibitor of pyruvate dehydrogenase kinase stimulates myocardial carbohydrate oxidation in diet-induced obesity: J Biol Chem, 2018; 293(25); 9604-13
101. Szczepaniak LS, Victor RG, Orci L, Unger RH, Forgotten but not gone: The rediscovery of fatty heart, the most common unrecognized disease in America: Circ Res, 2007; 101(8); 759-67
102. Bozkurt B, Aguilar D, Deswal A, Contributory risk and management of comorbidities of hypertension, obesity, diabetes mellitus, hyperlipidemia, and metabolic syndrome in chronic heart failure: A scientific statement from the American Heart Association: Circulation, 2016; 134(23); e535-e78
103. Libby P, Buring JE, Badimon L, Atherosclerosis: Nat Rev Dis Primers, 2019; 5(1); 56
Figures
Figure 1. Pathway diagram of hyperlipidemia leading to inflammatory response and oxidative stress. LDL – low density lipoprotein; FFA – free fatty acid; ROS – reactive oxygen species; SRC – tyrosine-protein kinase Src; RAS – GTPase HRas; ERK – mitogen-activated protein kinase; P38 – P38 mitogen-activated protein kinase; VAV – guanine nucleotide exchange factor VAV; Rac – Ras-related C3 botulinum toxin substrate; Lyn – tyrosine-proteon kinase Lyn; TXN – thierodexin; GSH – glutathione; oxLDL – oxidized low density lipoprotein; LOX-1 – lectin-like oxidized low-density lipoprotein receptor-1; RAGE – advanced glycation end product receptor; CD36 – CD36 antigen; LDLR – LDL receptor; PPARs – peroxisome proliferators-activated receptors; HO-1 – heme oxygenate-1; NQO-1 – quinine oxidoreductase 1; NF-κB – nuclear factor-κB; Nrf2 – nuclear factor erythroid 2-related factor 2; TNF-α – tumour necrosis factor alpha; IL – interleukin.Figure 2. Pathway diagram of myocardial endoplasmic reticulum and mitochondria damage caused by hyperlipidemia. FFA – free fatty acid; ROS – reactive oxygen species; JNK – c-Jun N-terminal kinase; ATP – adenosine triphosphate; ER – endoplasmic reticulum; DRP1 – dynamin-related protein 1; AC – acetyl group; Bip – binding immunoglobulin protein; P – phosphoryl group; oxLDL – oxidized low density lipoprotein; PERK – double-stranded RNA-dependent protein kinase-like ER kinase; eIF2 – eukaryotic initation factor 2; IRE1 – inositol-requiring enzyme 1; Bcl-2 – B-cell lymphoma-2; CHOP – the C/EBP homologous protein; PPARs – peroxisome proliferators-activated receptors; NF-κB – nuclear factor-κB. In Press
05 Mar 2024 : Clinical Research
Role of Critical Shoulder Angle in Degenerative Type Rotator Cuff Tears: A Turkish Cohort StudyMed Sci Monit In Press; DOI: 10.12659/MSM.943703
06 Mar 2024 : Clinical Research
Comparison of Outcomes between Single-Level and Double-Level Corpectomy in Thoracolumbar Reconstruction: A ...Med Sci Monit In Press; DOI: 10.12659/MSM.943797
21 Mar 2024 : Meta-Analysis
Economic Evaluation of COVID-19 Screening Tests and Surveillance Strategies in Low-Income, Middle-Income, a...Med Sci Monit In Press; DOI: 10.12659/MSM.943863
10 Apr 2024 : Clinical Research
Predicting Acute Cardiovascular Complications in COVID-19: Insights from a Specialized Cardiac Referral Dep...Med Sci Monit In Press; DOI: 10.12659/MSM.942612
Most Viewed Current Articles
17 Jan 2024 : Review article
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
14 Dec 2022 : Clinical Research
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387
01 Jan 2022 : Editorial
Editorial: Current Status of Oral Antiviral Drug Treatments for SARS-CoV-2 Infection in Non-Hospitalized Pa...DOI :10.12659/MSM.935952
Med Sci Monit 2022; 28:e935952